BIOL 2281 Lab Final Exam
1/87
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
88 Terms
Describe the basic process of an ELISA.
ELISA = Enzyme-Linked Immunosorbent Assay
Relies on antibodies to detect the presence of antigens in liquid samples
(1) Add samples to wells (positive control, negative control, infected, uninfected)
(2)Allow proteins to bind to the plastic (hydrophobic interaction)
WASH 2x
(3) Add 1' antibodies
WASH 2x
(4) Add 2' antibodies (attached by enzyme)
WASH 2x
(5) Add substrate (to detect presence of enzyme-labeled 2' antibodies)
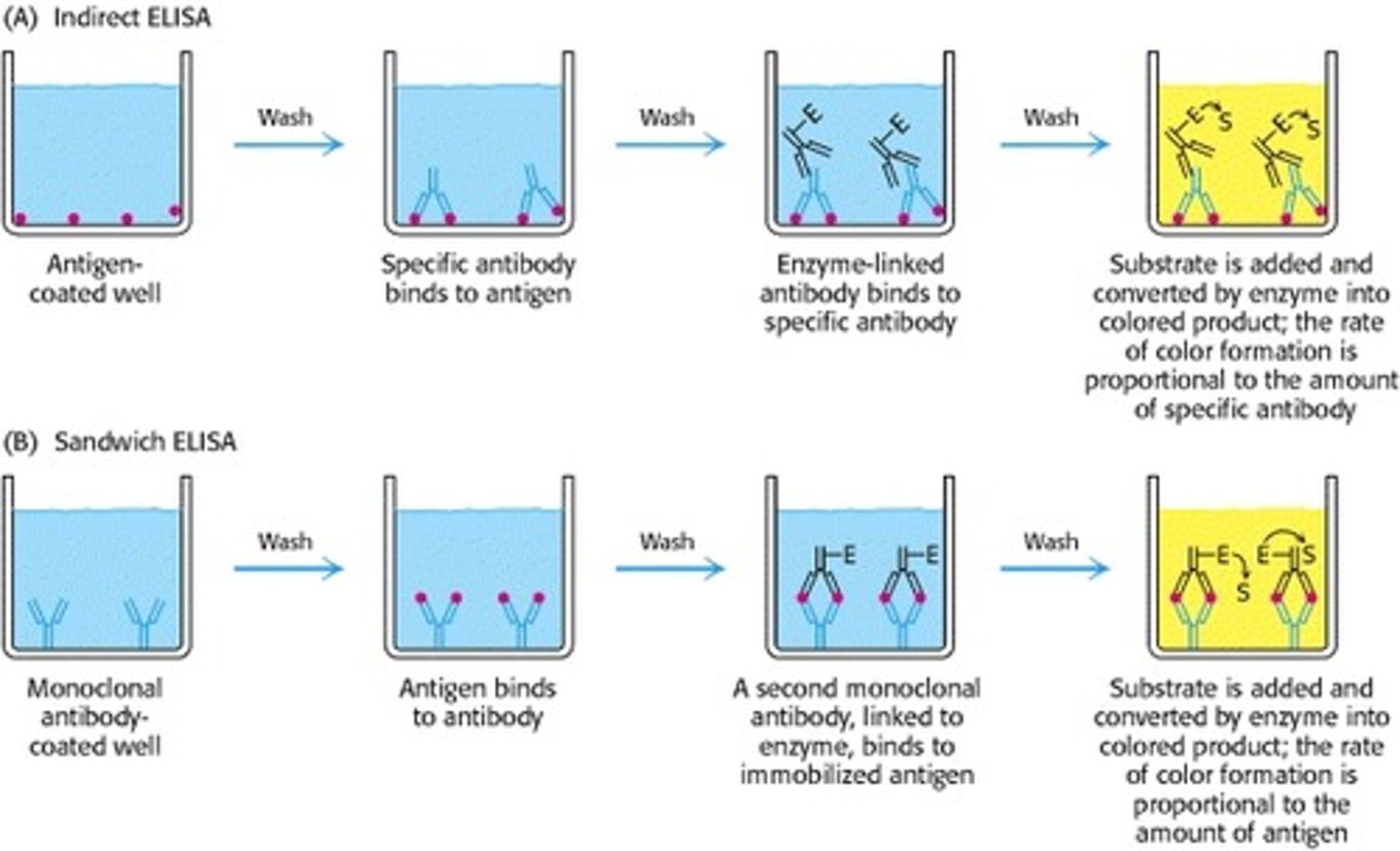
When you get a positive or negative result from an ELISA, what are you actually seeing?
Positive result: blue color, substrate have bounded to enzyme on 2' antibody, which is bounded to 1' primary antibody, which is bounded to antigen
Native result: no color, no reaction occurred because the substrate did not react with an enzyme; no enzyme-linked 2' antibody presence, it has already been washed off
What are the negative and positive controls? And what are their functions?
Positive control: To show that your assay ran correctly; in this case, would be the solution containing the antigen -- really we are testing for the enzymatic reaction
Negative control: To show what a negative result looks like; in this case, would be the buffer not containing the antigen
Given wells containing tested samples, be able to classify them as positive or negative results.
Blue wells = positive
clear wells = negative
What would happen if we DON'T wash after.. (3 scenarios)
1. don't wash after antigen: nothing happens because negative control has noa....
What cause false positive results?
What cause false negative results?
What is the purpose of a PCR reaction?
Amplify (reproduce) unique DNA regions to be detected in large genome → the amplified sample then allows for size determination, restriction mapping and nucleotide sequencing
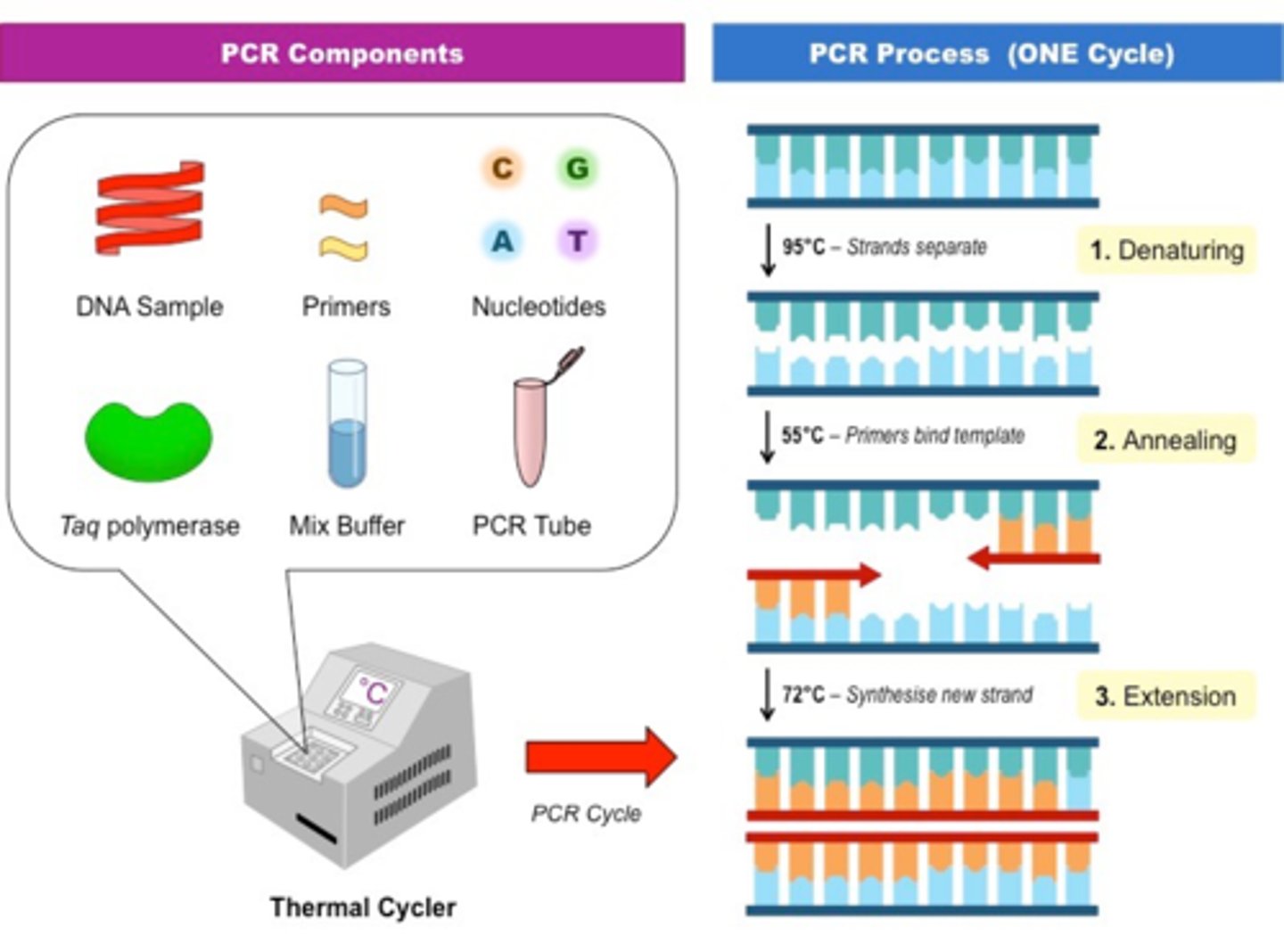
List essential components of PCR reaction (5):
1. Target mitochondrial DNA
2. Pair of primers for above DNA
3. Free deoxynucleotides to replicate, and as energy source
4. Heat stable DNA Tag polymerase
5. Buffer (contain cofactors and Mg+)
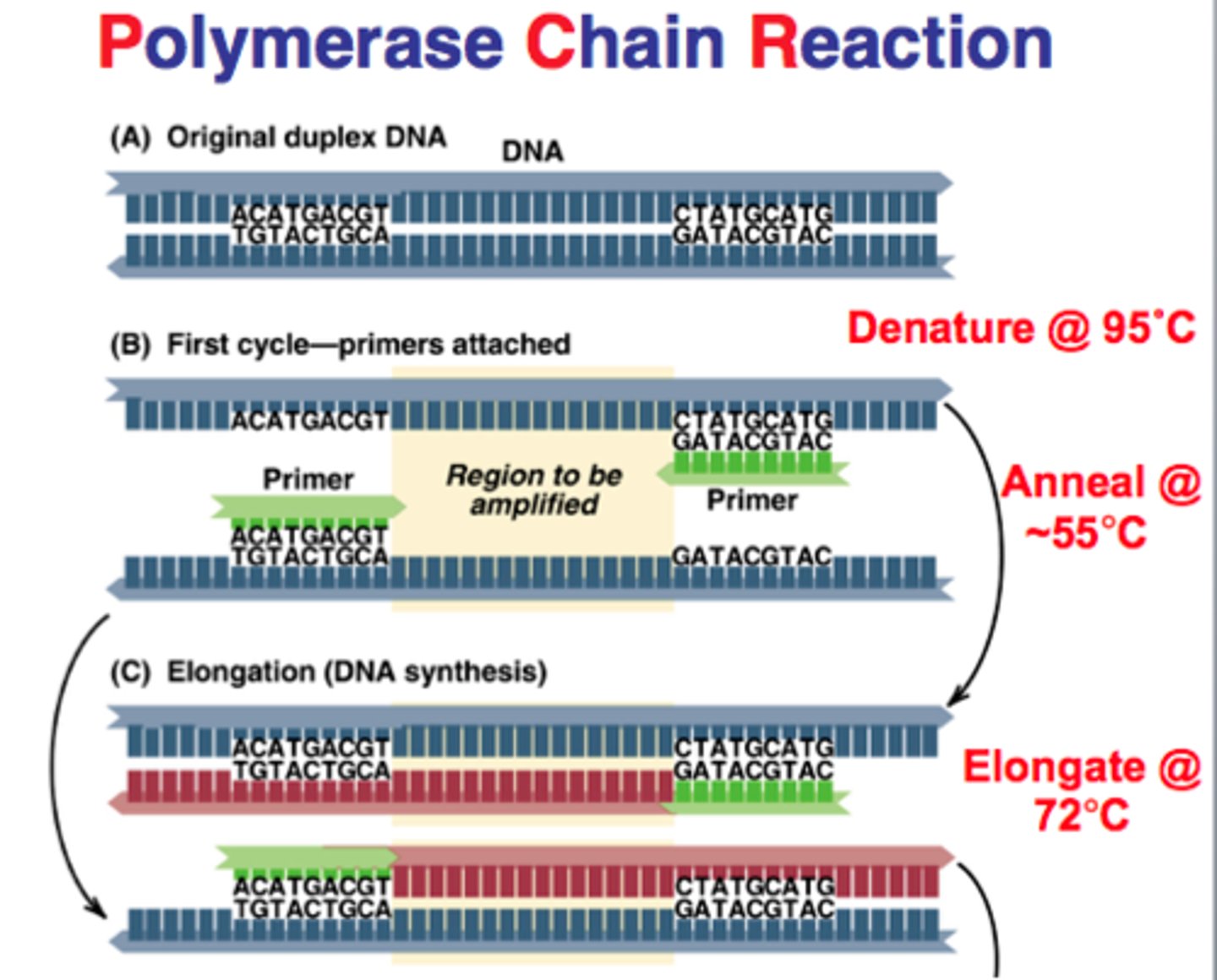
What kind of DNA can be used as template? Does it have to be pure?
Mitochondrial DNA, doesn't have to pure, can be crude extract as well. Reason being primers are specific to target DNA
The basic properties of PCR primers
Complementary to the 3' end of the gene
Specific to target DNA
Large, in excess, ~20 nucleotides long
Why should there be primers in the reactions?
DNA Polymerase cannot start replicating a DNA strand without the presence of a primer. Primase makes the primer.
What is special about the polymerizing enzyme used in the reaction?
Tag polymerase isolated from Thermos Aquaticus, thus is heat stable → allow to work in high temperature and survive heat shocks
What are the three basic steps in each cycle? The temperature used and the purpose of each step:
1. Denaturation: heating to ~90C, two separate DNA strands
2. Annealing: cooling to ~50C, allow primers to anneal to DNA
3. Extension: raising temp to ~75C, allow Tag DNA Polymerase to replicate
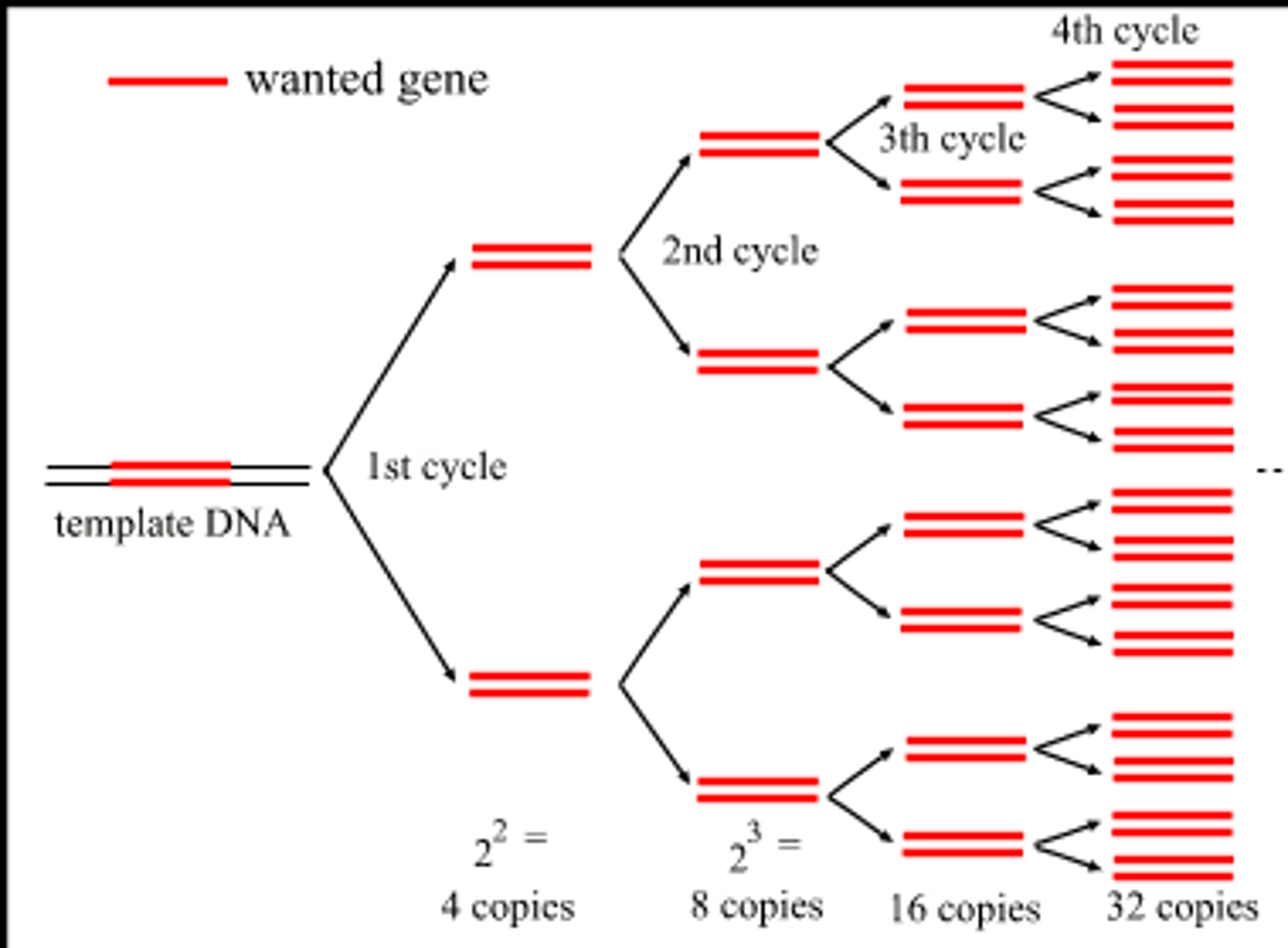
Is the amplification linear or exponential?
Exponential. Uses 2^n to determine the amount of DNA replicated available after n number of cycles
What determines the size of a PCR product?
The location of the primers (big number - small number)
What are the two functions of the dNTPs?
(1) Building block DNA
(2) Provide energy for reaction
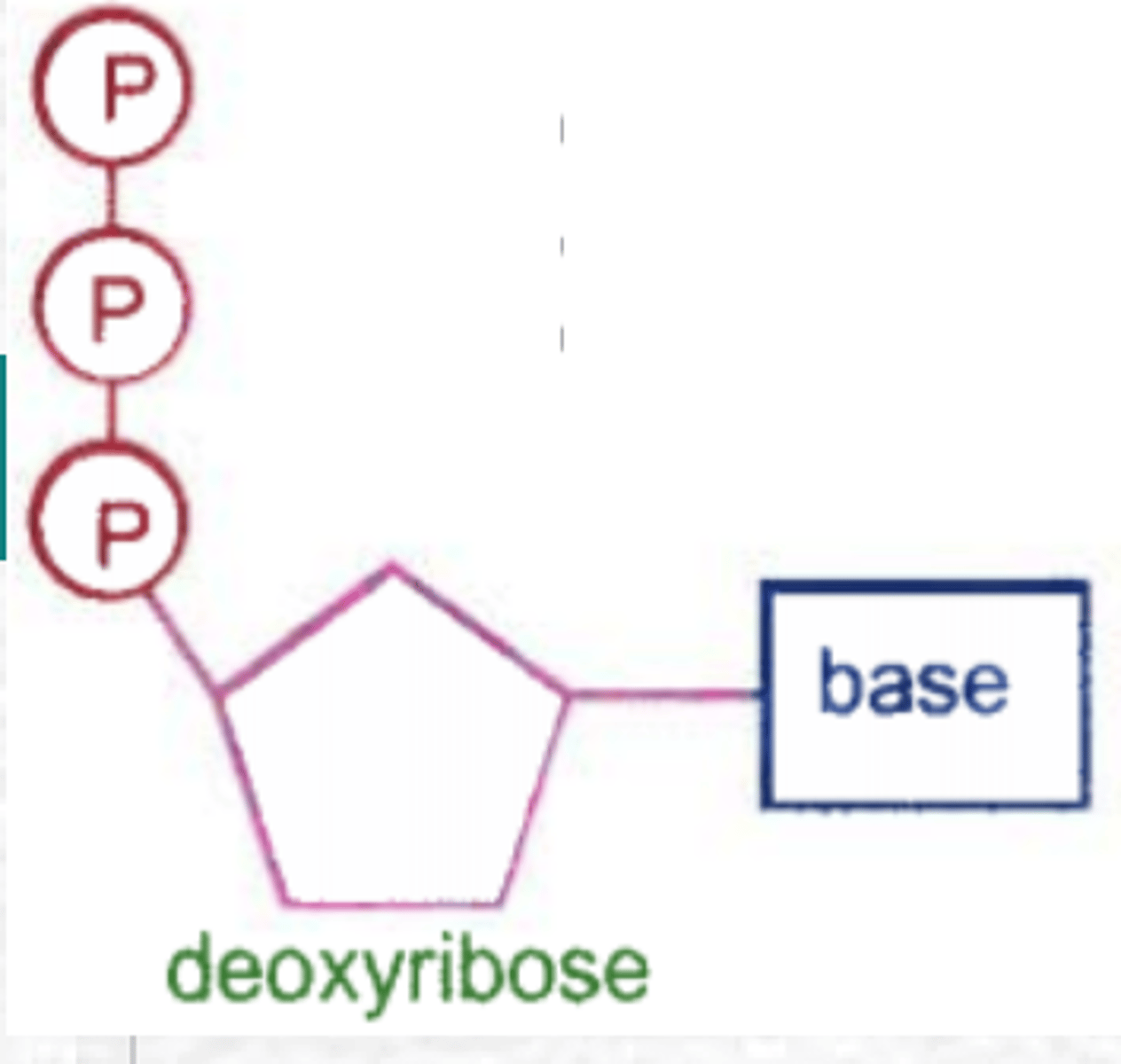
Why is MgCl2 included in the reaction?
(1) Acts as a cofactor (catalyst).
(2) Has a + charge while DNA has - charge --> MgCl2 neutralizes and act as as a buffer
(3) Enhances DNA amplification- increases the activity of Taq DNA polymerase, act as a source of magnesium
How to set up your PCR reaction in the lab
Part I: DNA Isolation
(1) Label 4 microcentrifuge tubes 1 → 4
(2) Pipette 100 ul of Chelex into m.tube 1, put aside
(3) Swish 10 ml of saline solution for 30 seconds
(4) Expel saline into cup → Swirl → Pipette 1.5 ml in tube 2 and 1.5 ml into tube 3
(5) Centrifuge tube 2 and 3 for two mins → will see supernatant layer and cell layer in tubes
(6) Pour off supernatant layer, leaving only cell layer in tubes
(7) Resuspend the cell layer by pipetting it up and down (mixing)
(8) Pipette 20ul of cell from tube 2 to tube 1; then 20ul of cell from tube 3 to tube 1
(9) Boil tube 1 for ten minutes in water bath (Boil DNA to denature the DNA)
(10) Centrifuge tube 1 for two minutes after boiling
(11) Pipette 20 ul of supernatant (contain DNA) from tube 1 to tube 4
Purpose of Chelex:
Complete removal of cofactors and PCR inhibitors from a solution such as contaminating metal ions that catalyze degradation of DNA --> prevent DNA degradation
Purpose of boiling
Lyse the cell/make the cell porous to release DNA strands and inactivate cellular proteins that may interfere with PCR reaction
A student performed a PCR reaction and he wants to know if he has obtained the expected PCR product. What does he need to do?
Determine PCR size before hand → do experiment → Run a gel electrophoresis, then compare with ladder on the gel (will show you how many pairs are in your product)
What is the basis of separation of DNA on an agarose gel? How is it different from the protein agarose gel (E11) in terms of the setup or the property of the gel?
DNA Agarose gel:
- reducing gel
- DNA separated by size
- E.b added
- comb on the black cathode On DNA agarose gel, DNA is separated by size of DNA fragments; component of gel is not a protein; has Ethidium bromide added, use UV light to see; place comb at the end on the black cathode
Protein agarose gel:
- native, nonreducing gel
- protein separated by net charge
On protein agarose gel, protein is separated by protein's net charge; component of gel is protein, can place comb in the middle, depends on pI and pH; don't use ladder for protein
What gives DNA the negative charge?
Phosphate groups in the DNA backbone carry negatively-charged oxygen molecules
What affect the migration rate of DNAs in agarose gel electrophoresis?
Sizes, shape of DNA fragments, agarose, concentration, buffer, voltage
(bigger < than smaller/supercoiled
What chemical "stains" the DNA in our experiment? And how?
Ethidium bromide intercalates between bases of a strand of nucleic acid. When excited by UV light, it resonates and gives off a fluorescent light.
What is the purpose of including a marker lane in the gel?
Marker lane (DNA ladder), contains DNA fragments of known size, give us something to which we can compare our PCR products in order to estimate the size of our fragment (in base pairs)
How does the size of linear DNA molecules relate to their migration rate?
Migration rate of DNA fragments is inversely proportional to the log of their weight. (small fragments move faster than large fragments)
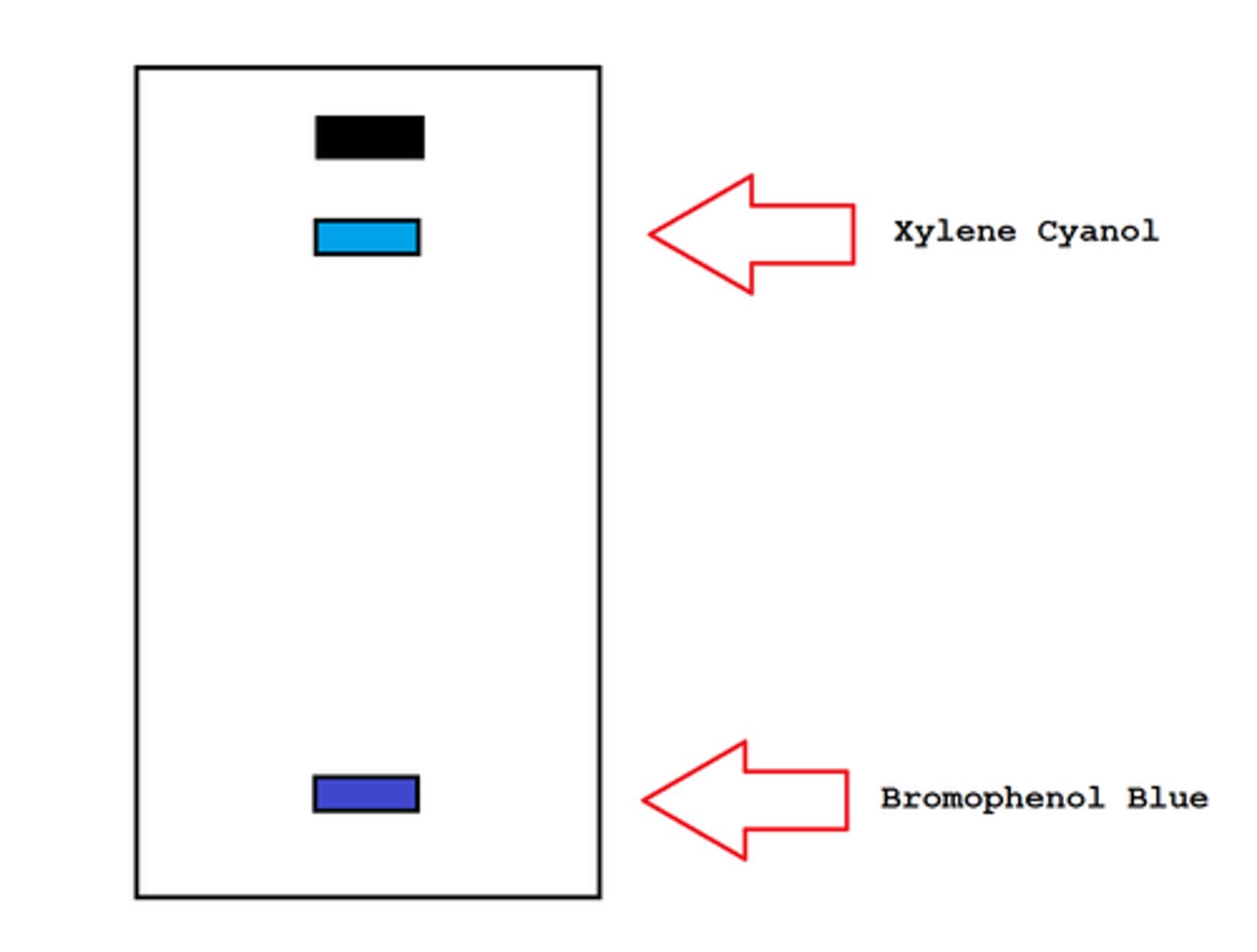
What two blue dyes are included in the marker? Why?
2 dyes are bromophenol blue and Xylene cyanol, and they migrate in the gel and allow visual monitoring of how far the electrophoresis has proceeded.
The "faster" dye, bromophenol blue, co-migrates with DNA fragments approximately 300bp
The "slower" dye, Xylene cyanol, comigrates with DNA fragments approximately 9k bp in size.
You amplified a segment of your own mitochondrial DNA. Do you expect the sequences of your PCR product vary from those of your classmates? Why or Why not?
The mitochondrial DNA itself will vary between students because each of us inherited it from our moms; but the sequences of our PCR product will be the same because the same region is being amplified.
Why does the PCR reaction catalyzed by the Taq DNA polymerase has a higher mutation rate than the normal DNA replication process found in living cells?
Has no proof-reading mechanism
What reagent is added to the 0.5X TBE buffer to allow visualization of the DNA bands in the agarose gel? Describe how you can see the DNA after the gel running is stopped
Ethidium Bromide is added; it fluorescent under UV light
Define genetic transformation:
Expression of genes that have been introduced into an organism → GMO plants, gene therapy where genetically transforming a sick person's cells with healthy copies of the defective genes that causes the disease
Define competent cells. How do we make competen cells in the lab?
cells made permeable to plasmid DNA using lab techniques.
How did we do that in lab?
Calcium chloride and heat shock
What are some other methods of making competent cells:
- Electroporation: using a pulse of electricity to briefly open the pores
- Protoplast formation: forming cells that don't have cell walls (removed by enzymes)
- Microprojectiles: uses gun to introduce particles into cells with high-velocity microprojectiles
Define Constitutive gene expression:
Transcribed continually, always ON, as opposed to facultative/repressive gene, which only transcribed when needed;
regulation is limited by metabolic state of organism, most of constitutive genes are housekeeping genes
Example of Constitutive gene expression in the lab:
Beta-lactamase, has to be on throughout the whole time, otherwise, won't be able to grow on ampicillin medium
Define inducible gene expression:
Expression can be either responsive inducible to environmental change or dependent on position in the cell cycle
Example of Inducible gene expression in the lab:
GFP system--- only turned on when Arabinose is present in growth medium
Important characteristics of the bacterial strain in this lab:
- a single celled bacteria,
- reproduce quickly,
- can't infect plants/animals,
- not natural antibiotic resistant thus can't grow on Lb/amp plates
Important characteristics of the plasmid (4) in this lab:
small non-chromosomal DNA molecules; pGLO, contain (4):
- Gene of interest (GFP)
- Gene of a selective advantage to the bacterium (bla or Ampr) → codes for beta-lactamase protein
- Origin of replication
- Gene regulation system for GFP (promoter Pbad, araC gene) - Polylinker area (multiple endonuclease cleave stites) ??
Understand how bacterial transformation is set up in the lab
Why do we use LB/amp media to select for transformants?
Because LB is a rich medium for growing bacteria, adding ampicillin provides a means of selecting transformants that have taken up plasmid DNA containing the bla gene, which encodes resistance to ampicillin. Consequently, only bacteria that have been successfully transformed with such a plasmid will survive in LB Amp.
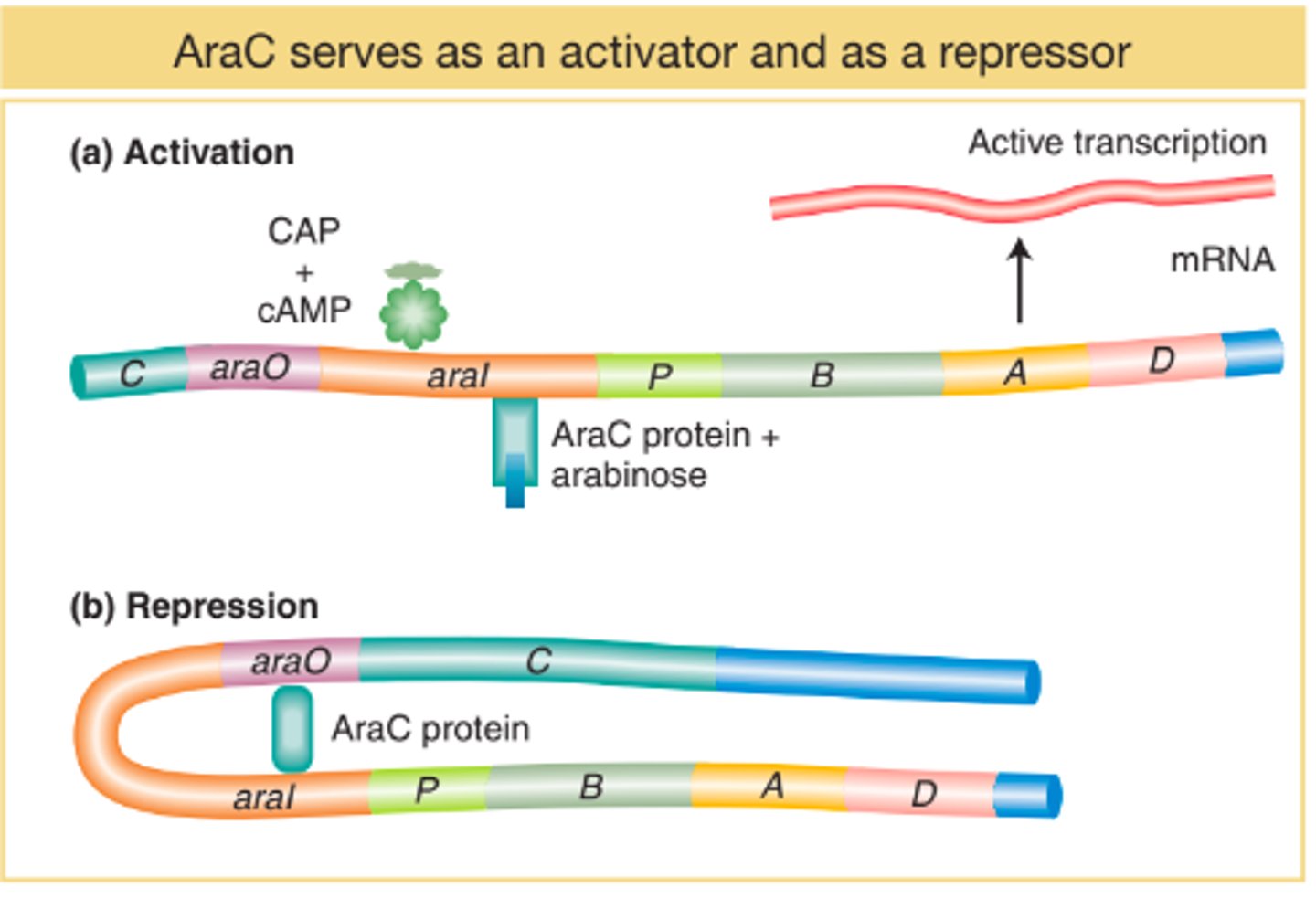
What was the purpose of arabinose
To activate expression of GFP gene = allowing the bacteria to glow under UV
Without arabinose, araC protein sits on promoter Pbad and polymerase can't get to promoter to start transcription of GFP gene
With arabinose, it binds to araC protein → araC protein change conformation → exposed promoter Pbad → polymerase can now start transcription of GFP gene
Steps to enhance transformation efficiency:
Incubation: cells mixed with CaCl2 (37 C) → make cell membrane porous
Heat Shock: quick temp changes (0 C → 42 C → 0 C) → to uptake the DNA you are trying to perform
Recovery: Short incubation
Use cells growing at log(Exponential) phase
Determine the transformation efficiency given the number of transformants, the concentration and volume of plasmid used.
Took pic look in photos
Arabinose operon
A cluster of genes (araB, arabC, arabA) controlled by promoter Pbad, DNA-binding protein araC
What does growth on each plate looks like? Why?
LB only: lawn
LB/amp: no growth (bc natural e coli does not have bacterial resistance)
LB/amp/+pGLO: colonies, bc some have survived
LB/amp/ara/+pGLO: colonies + glow bc of arabinose
What allows the transformed cells to grow on LB/amp? How?
pGLO, it contains the genes that when expressed produce the beta lactamase enzyme which gives resistance to ampicillin
What might cause growth on the negative control plate?
- Contamination from a bacteria that is resistant
- not changing pipette tips
- utilizing the wrong growth mediums
What are the control plates? What are their purposes?
Negative control: Lb/amp plates without the pGLO; purpose is to show that the result would be without pGLO added → result: no growth
Positive control: Lb plate without pGLO → result: TMTC ??
How does parallel dilutions different from serial dilution?
Serial dilution: method of diluting a stock solution where concentration decreases by the same quantity in each successive step
Parallel dilution: done to make more than one dilution where desired concentration are unrelated to each other
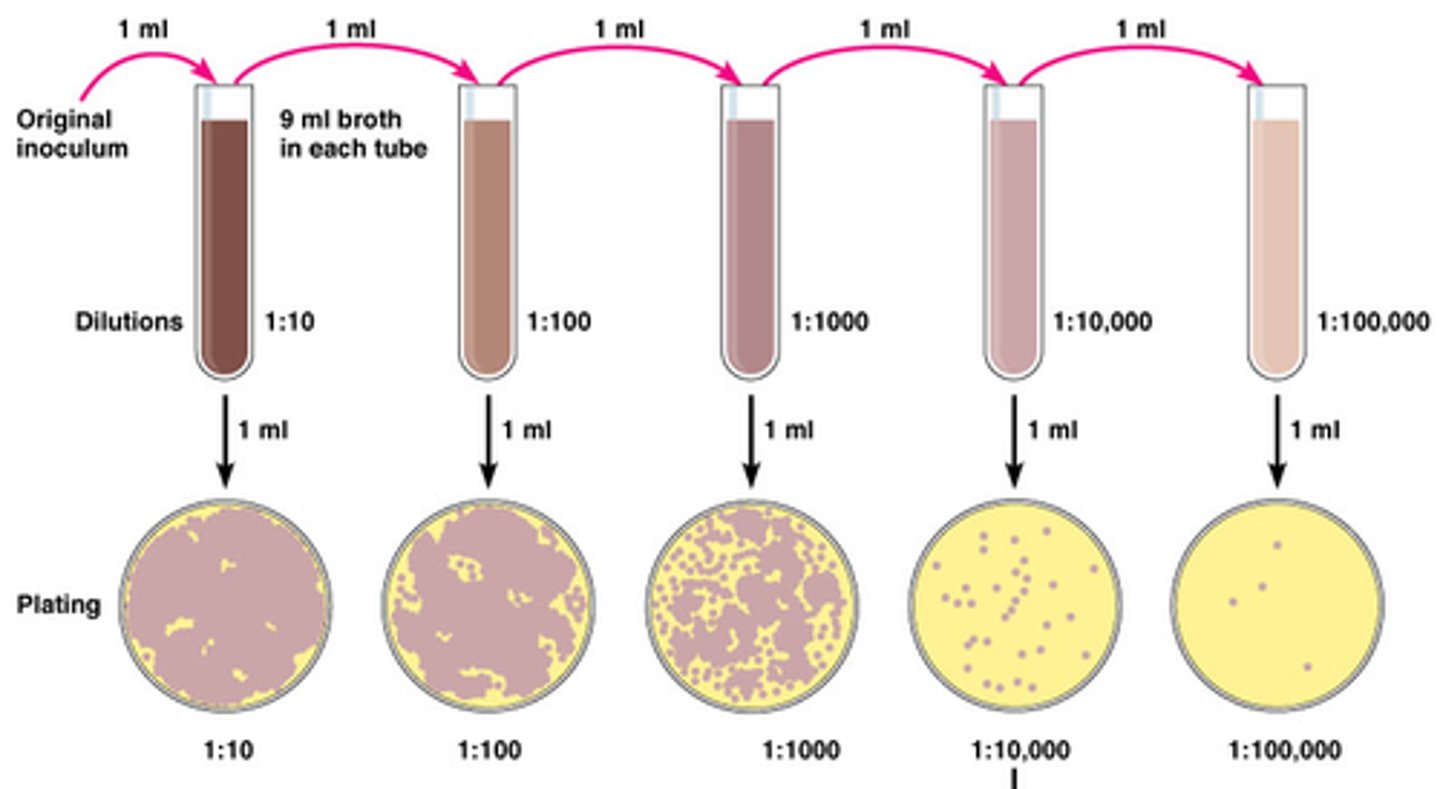
How to prepare a parallel dilution method?
Use C1V1 = C2V2 where C1 and V1 are concentration and volume of stock solutions, C2 and V2 are concentration and volume of dilute solution
What is spectrophotometry
method to see how materials absorb light by measuring the intensity of light
What is an absorbance spectrum?
How is it generated?
Graph? (title, axis, units).
Spectrum of electromag radiation transmitted through a substance, showing dark lines or bands due to absorption of a specific wavelengths
Generated by measuring absorbance at different wavelength
(absorbance vs wavelength)
Title: X product absorbance spectrum
X-axis: Wavelength (nm)
Y-axis: Absorbance (A units)
Linear
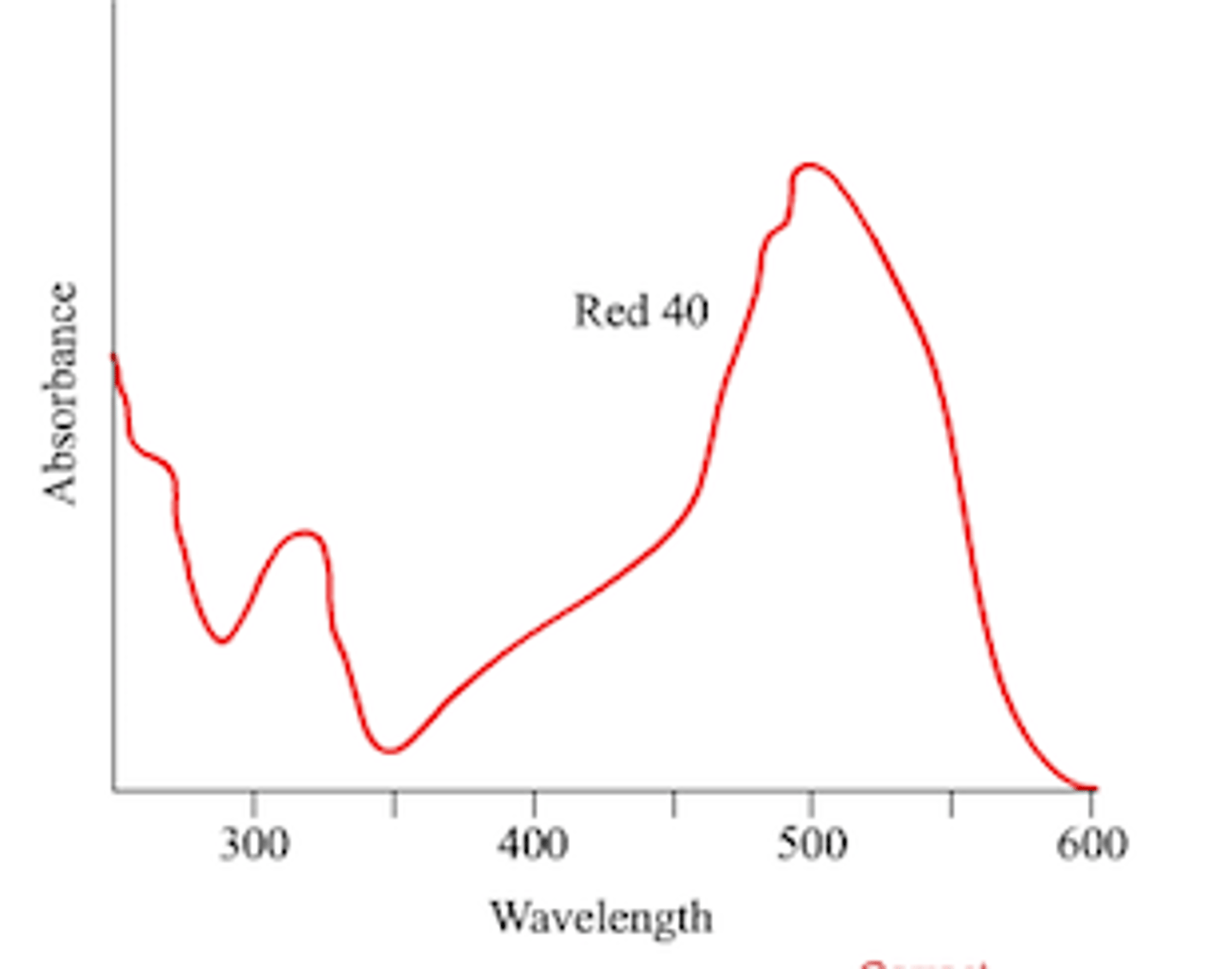
Define lmax and how is it determined?
Wavelength where maximum absorbance occurs; experimentally determined by taking a series of readings at different wavelength, then find the two wavelengths (before and after) near the highest absorbance, then add and divided by 2
What is the purpose of the standard curve?
A graph showing a chemical's absorbance at its wavelength max vs. its concentration; linear relationship; can be used to determine the concentration of an unknown substance by measuring absorbance in a spec, then find the corresponding concentration on the standard curve
Why such a curve is typically measured at the lmax of a chemical?
Because this is the wavelength where maximum absorbance occurs for the chemical whose concentration is being measured. This allows us to distinguish the chemical in solution from other chemicals that are similar.
Be able to describe the Beer's law and relate it to the standard curve
A= bec
A: Absorbance
b: pathlength (cm)
e: molar absorptivity (Lmol-cm-)
c: molar concentration molL-
Know how to calculate e giving Beer's Law
Spectrophotometer:
1. Turn on visible light
2. Autozero the spec
3. Setting specific wavelength
4. Measure absorbance of wavelength
Be able to turn on the visible light and autozero the spectrophotometer, setting a specific wavelength and then accurately measure the absorbance of a solution at that wavelength.
watch vid
What may influence the activity of an enzyme? List at least 4 factors.
Temperature:
Optimal is 20-40 C, up to 45 C, Denatured/inactivated above 50-70 C,
pH: most optimal at neutral pH; acid phosphatase is at pH 4.5
Amount of enzyme,
Substrate concentration, effects of inhibitors/activators
Describe how the amount of enzyme affects the rate of a reaction.
Increasing enzyme → more active sites available → increase reaction rate
Describe how the amount of substrate affects the rate of a reaction.
Increase substrate → increase reaction rate until all active sites are saturated → reaction rate levels off
Rate of reaction is limited by the speed which substrate is being converted into product
Understand why reaction rates decline with time and use this information to correctly process the data (by choosing the proper data points to do linear regression)
- because substrate or cofactors become depleted, breakdown of enzyme
Exclude 0,0 and the first point of level off line. Choose two points from the linear portion of the graph to get the slope (this is the reaction rate- nmoles of product formed over unit time). Do not use points from the flat portion of the graph where the amount of product formed is no longer increasing.
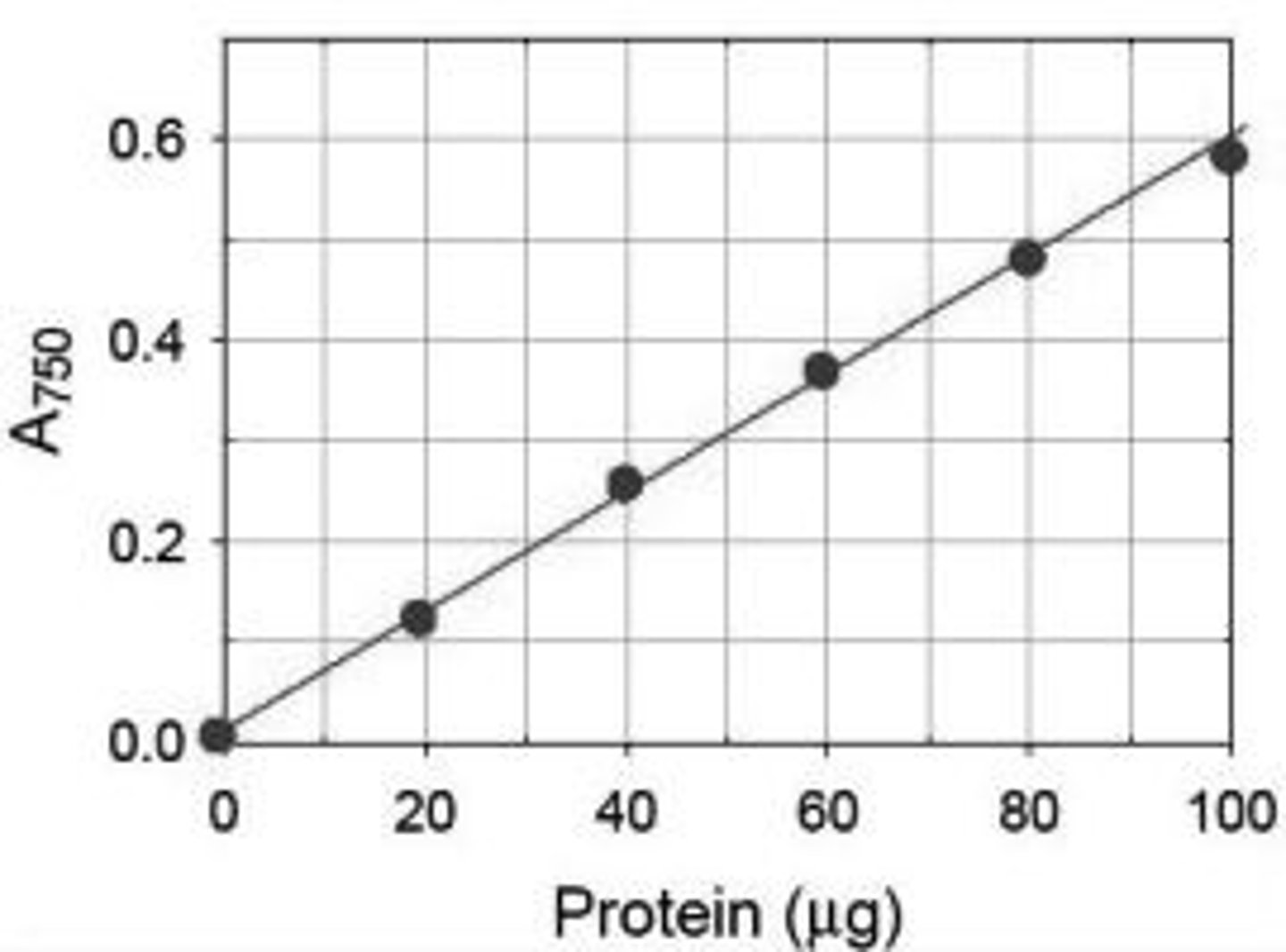
The standard curve of absorbance vs nmole/ml of nitrophenol
The standard curve is constructed by taking the absorbance of a set of solutions of known concentrations. The absorbance is then plotted in a graph against concentration of nitrophenol. We use a spectrophotometer to determine absorbance. We use a blank, or a solution containing all the components of the solvent except for the solute we are measuring the concentration of to autozero. This is useful because it enables us to determine the concentration of an unknown given the absorbance and the equation of the line of the standard curve.
The amount of nitrophenol produced in the assay vs time
This graph is constructed by removing a portion of the main solution containing the enzyme reaction, placing it in a separate test tube, and adding KOH (or some other chemical depending on the type of enzyme) to denature and stop the reaction at various time intervals. These various solutions are then placed in a spectrophotometer and the absorbance is determined. Using the absorbance, the concentration of nitrophenol (product of the enzyme assay) can be determined, and amount in nmoles can be found. This graph of nmoles of nitrophenol vs time allows us to determine the reaction rate of the enzyme (the slope of the graph)
Be able to determine the Vo given the appropriate graph and equation.
Vo is slope of reaction rate graph
Be able to describe the reaction catalyzed by acid phosphatase and describe how the assay was performed, and the functions of KOH in the assay
Acid phosphatase cleaves a phosphate group from NitrophenYL phosphate (colorless) to produce phosphate groups and NitrophenOL. NitrophenOL reacts with an alkaline solution (KOH) and turns yellow. KOH also stops the reaction
Functions of KOH in the assay:
Stops the reaction (enzyme needs acidic condition)
Turns the nitrophenol yellow
Be able to label amino group, carboxyl group and side group
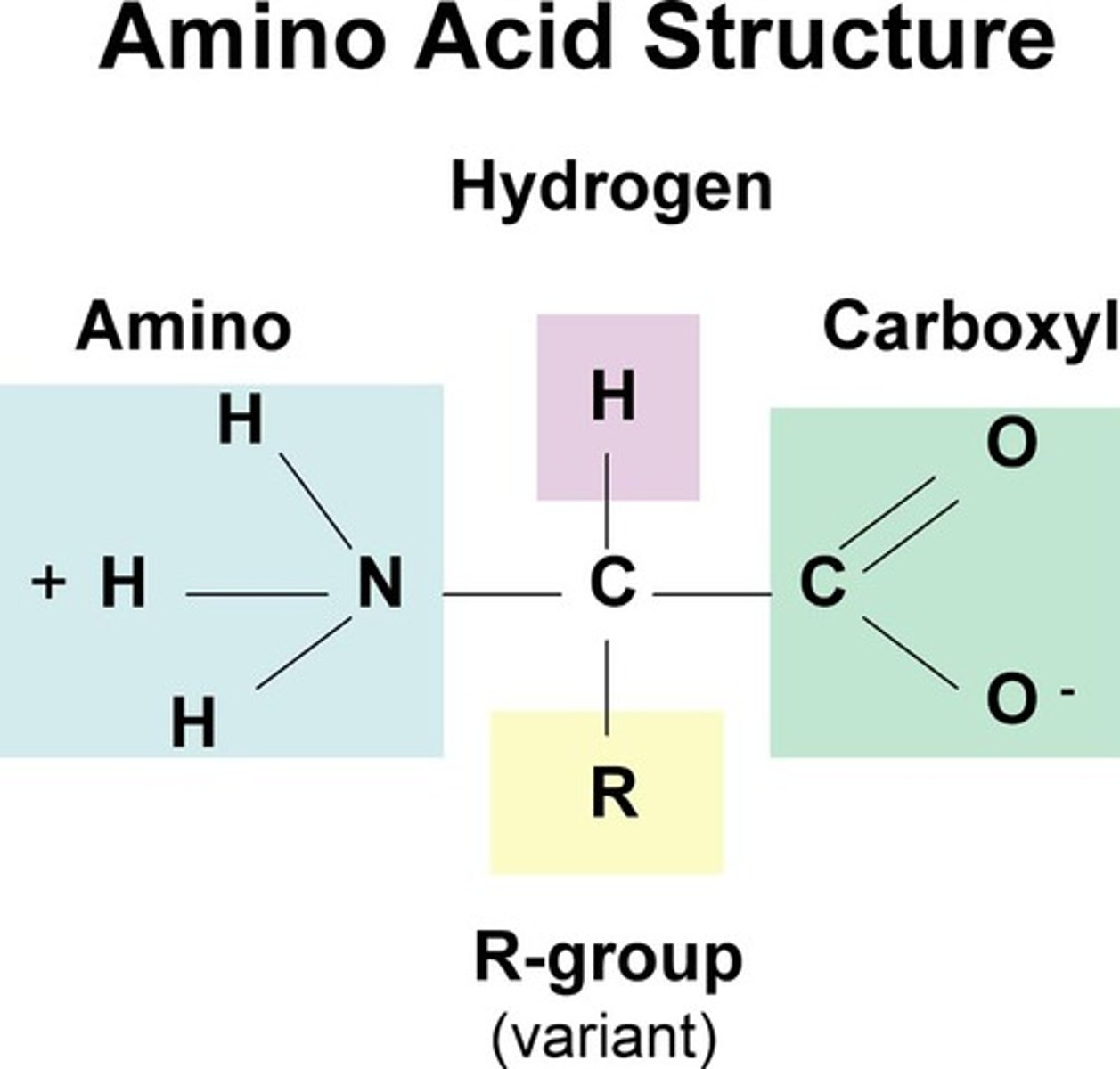
Provided with the structure of amino acids, be able to classify them into non-polar, polar or charged amino acids
What are the 3 basic/positively charged amino acids?
Histidine His H
Arginine Arg R
lysing Lys K
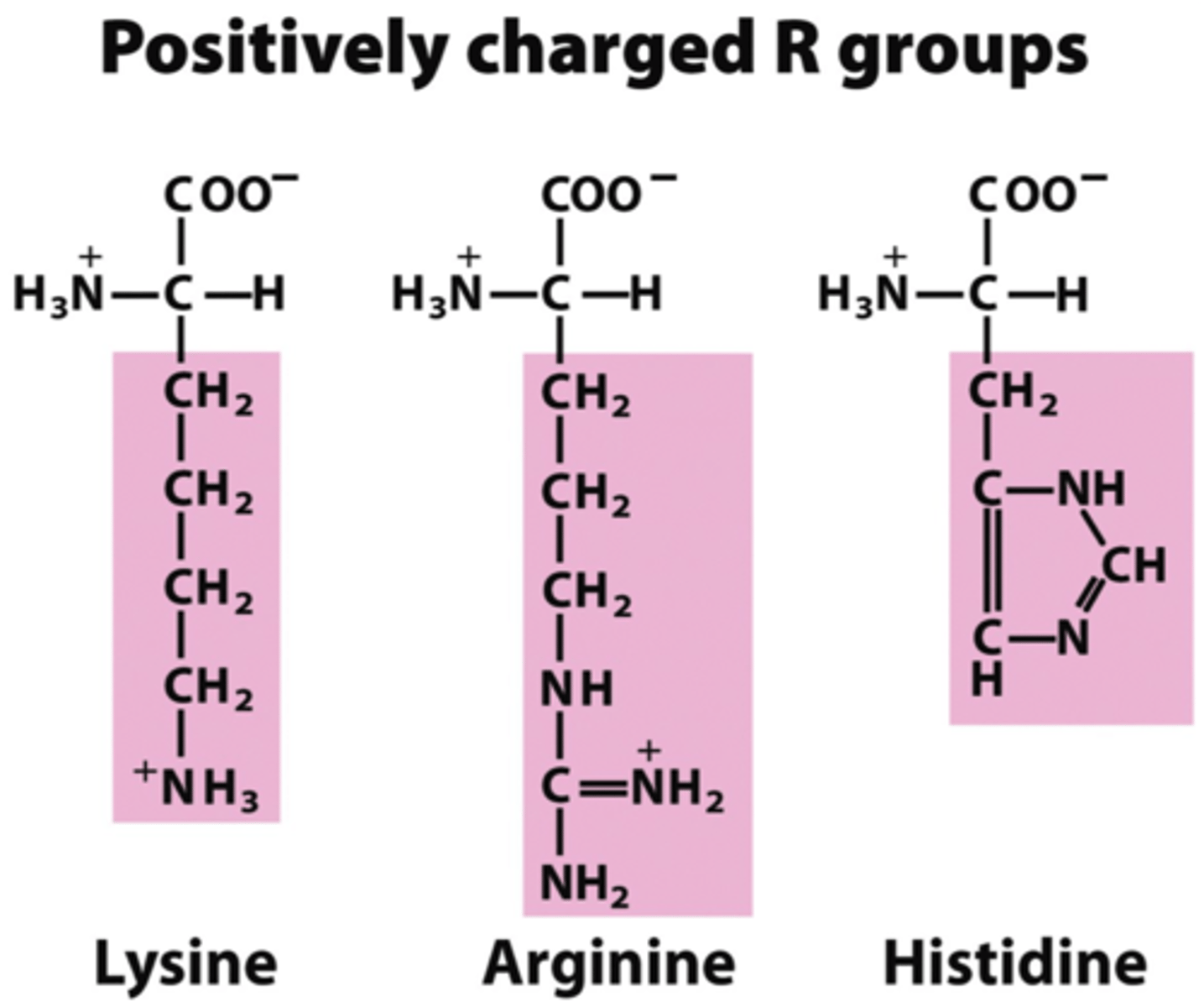
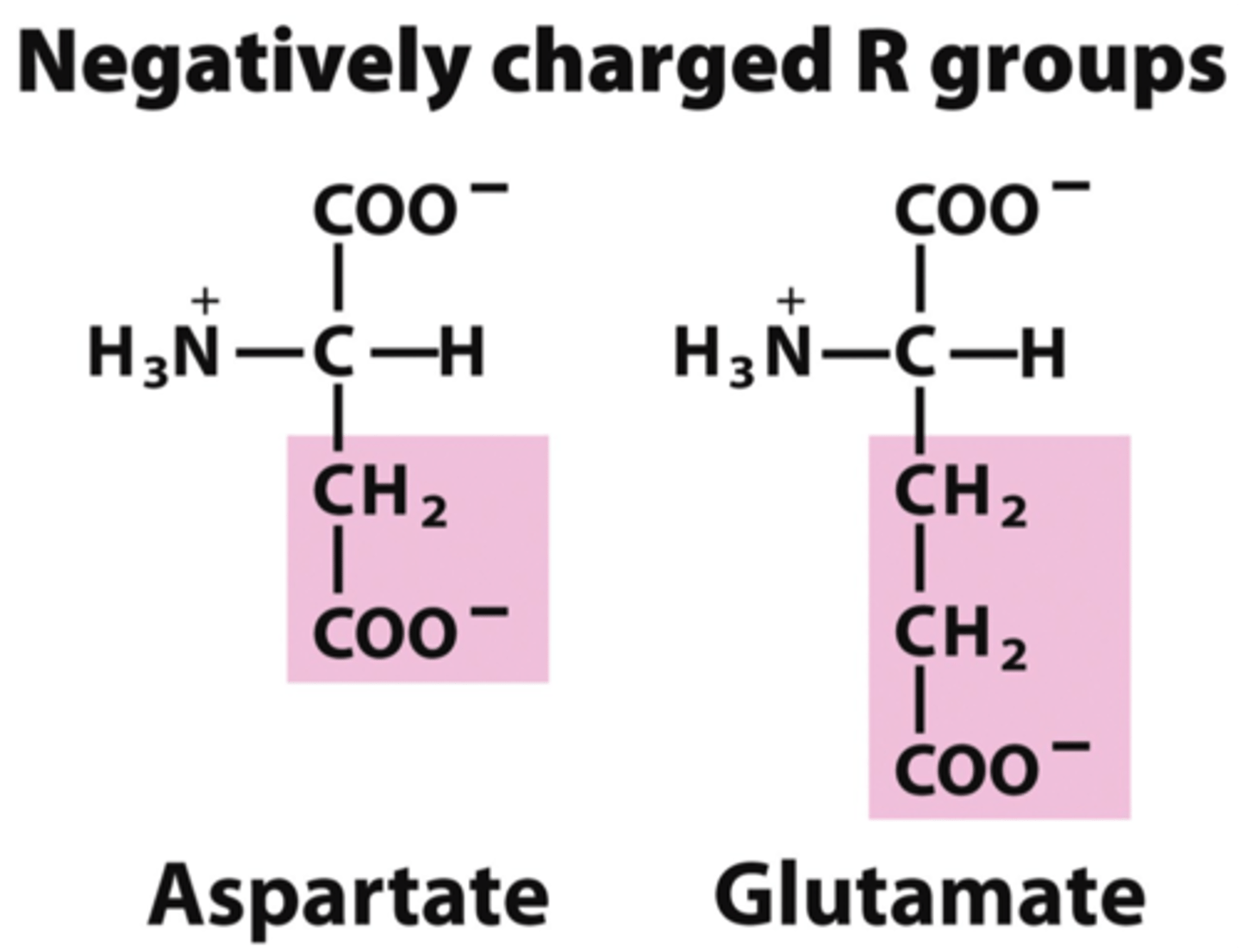
What are the 2 acidic/negatively charged amino acids?
Aspartic acid Asp D
Glutamic acid Glu E
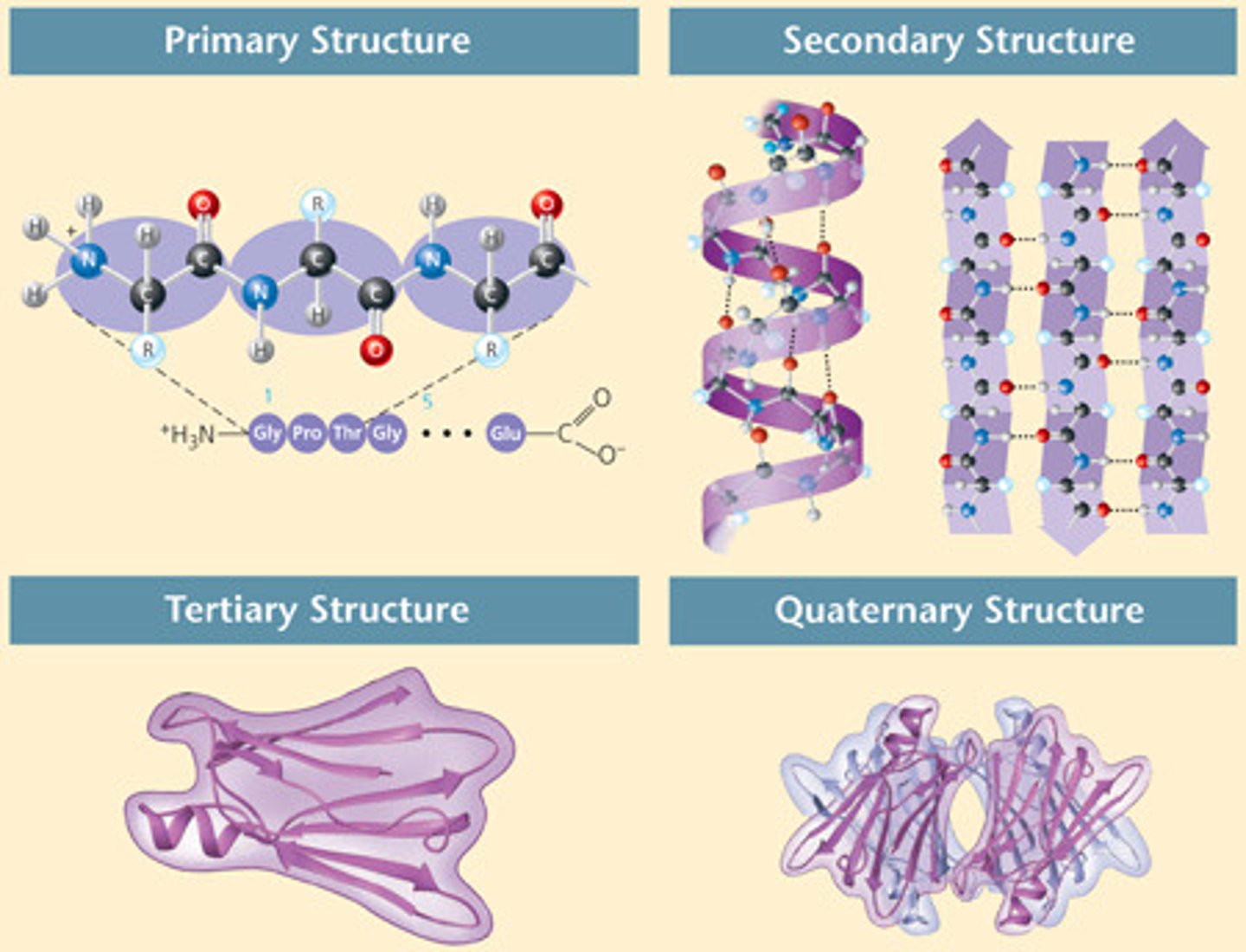
Define the four levels of protein structures.
1. Primary: amino acid
2. Secondary: 3D form of local segments, form H bonds, either alpha-helices or beta-sheets
3. Tertiary: 3D shape via R group interactions
4. Quarternary: only for proteins composed of >2 polypeptide chains
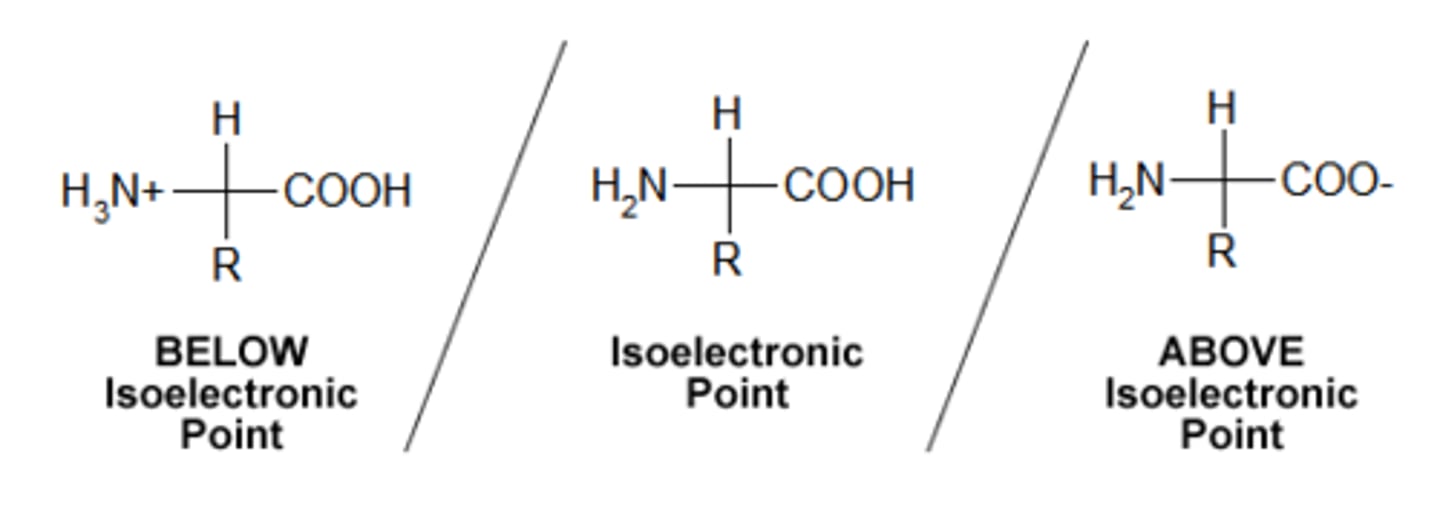
Define pI
Specific pH of solution where the protein's net charge is zero.
Know the relationship between acidity of amino acid or proteins and pI
if there are more acidic aa than basic aa, then the protein is more acidic and thus negatively charged → requires more H+ to be protonated → lower pH solution to be neutral → lower pI → pI is lower than 7
If there are more basic aa than acidic aa, then the protein is more basic, thus more positively charged → requires less H+ to be protonated → higher pH solution to be neutral → higher pI → pI is higher than 7
What would the protein charge be if pH > pI? What electrode will protein migrate to?
What would the protein charge be if pH < pI? What electrode will protein migrate to?
If pH solution is > than pI → excess H+ in solution → amino & carboxyl group protonated ( NH2 → NH3+ ; COO- → COOH) → amino acid is positive charged
If protein positively charged (+) will migrate → to black cathode (-) pI > pH, cations toward cathodes
If pH solution is < than pI → excess OH- in solution → amino & carboxyl group deprotonated ( NH3+ → NH2 ; COOH→ COO-) → amino acid is negatively charged
If protein negatively charged (-) will migrate → to red anode (+) pI < PH, anions toward anodes
Understand the difference between normal hemoglobin and mutated hemoglobin in sickle cell patients;
Sickle cell anemia is caused by a single based amino-acid mutation in the gene that expresses hemoglobin protein. In mutated hemoglobin, Glycine 6 (polar) is replaced by Valine 6 (nonpolar) on the beta chain → caused a change in net charge, thus conformation of protein → When hemo S is deoxygenated, it crystallizes in red blood cells and this lead to distortion of red cells into a sickle shape → abnormal cells are destroyed rapidly in the body, → reduced erythrocytes → anemic
Valine 6 (non polar) replaces Glycine 6 (polar) in beta chains of Hemo S → Hemo S has 2 fewer negative charges per hemo mole → Hemo S are more basic → has a higher pI level than 6.9 → migrate slower toward the anode;
be able to compare the migration rates of the two types of hemoglobin (HbA and HbS) based on the nature of the mutation.
Hemo A (pI 6.9, more acidic, negative charged) migrates further towards anode
Inherited as autosomal recessive gene → if patient is heterozygous (HbAHbS), then won't express it, only carry the trait = half of circulating hemoglobin is Hemo S, half is Hemo A; rarely anemic
Understand the separation principle of protein agarose gels (native or non-reducing gel electrophoresis)
Net charges.
What determines the rate of movement of the proteins?
Different between pI of protein and pH of buffer.
Greater difference = faster migration
Does the molecular weight of a protein affect its migration?
No
Does the separation require proteins to be denatured?
No, because we are using native/non-reducing gel electrophoresis
Does the protein change its shape while it is moving in the gel
No
Electrophoresis:
separation technique where electrical field causes charged molecules to move through a gel
Properly load a sample into an agarose gel. Assemble the gel apparatus and connect to the power supply correctly (the wells was set at the end of the gel).
watch vid