Hematology/Oncology
1/227
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
228 Terms
Sarcoma means a tumor of __ origin
-mesenchyme
Angiosarcoma
-break it up
-appearance?
-good prognosis or bad?
-common occurrence locations? Note their associated causes
-arise from what skin layer?
-angio is blood vessel, sarcoma is a tumor of the mesenchyme; it is a tumor of the vessels and lymph channels
-purple nodule/plaque
-bad
-breast(radiation), liver(vinyl chloride exposure), beneath the skin around head and neck(sun exposure)
-from the dermis(this is where blood vessels are!)
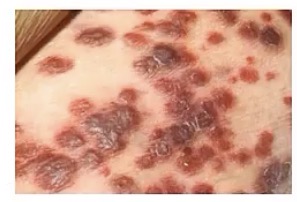
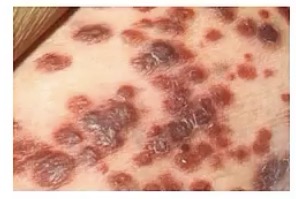
Bacillary Angiomatosis
-zoonotic infection by __
-common in patients with __, meaning?
-this will move systemically through __
-appearance?
-very similar to Kaposi sarcoma, so note the presence of what cell types? How is this different from Kaposi sarcoma?
-Bartonella
-HIV/AIDS; they’re immunocompromised
-blood vessels
-numerous red/purple nodules
-lymphocytes AND neutrophils; only has lymphocytes
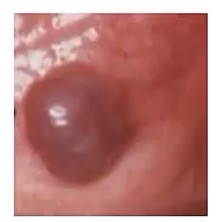
Kaposi Sarcoma
-common in patients with __
-diagnostic indicator is they are positive for __
-very similar to Bacillary Angiomatosis, so note the above, as well as presence of what cell types? How is this different from BA?
-HIV/AIDS
-HHV-8
-ONLY lymphocytes; has lymphocytes AND neutrophils
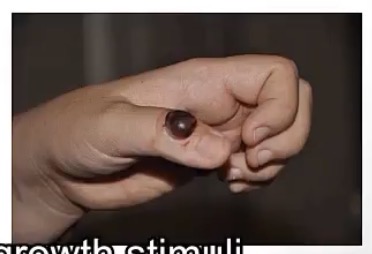
Pyogenic Granuloma
-generally describe
-what are the causes that trigger what’s mentioned above?
-common characteristic?
-how are they treated?
-vascular hyperplasia due to “growth stimuli”
-pregnancy and trauma
-bleed profusely
-surgically removed
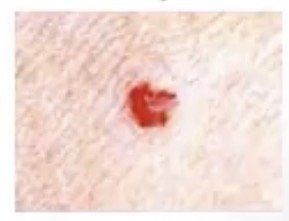
Cherry Hemangioma
-common in what age group? Why?
-appearance?
-middle-aged to elderly; develop with aging
-multiple small red blots, classically on the trunk
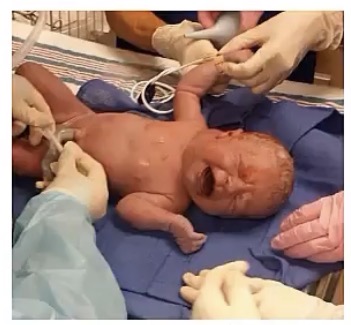
Cystic Hygroma
-present in what age group? Why?
-caused by obstruction of __, thus filled with __
-classically appear where?
-can be identified prior to birth using what scan?
-while they themselves are benign, why is their appearance a concern?
-newborns; they’re congenital
-lymph drainage; lymph
-the neck
-prenatal US
-they’re associated with higher risk of aneuploidy and malformation, such as Down or Turner syndrome
Glomus body
-what are they? Where are they?
-function? How?
-found in what skin layer?
-complication of these?
-modified smooth muscle cells; in the fingers and toes
-preserved heat; shunting blood away from surface in cold
-dermis of the skin
-glomus tumor
Glomus tumor
-what are they?
-where?
-normal function of glomus bodies?
-how will these present?
-modified smooth muscle cells
-in the fingers and toes
-preserve heat by shunting blood away from the surface, in the cold
-as pink/purple nodules that are painful when exposed to the cold
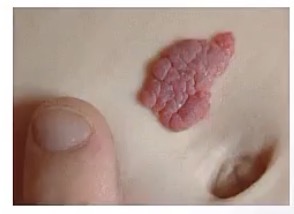
Strawberry Hemangioma
-what is it?
-appear in what age group? At birth?
-presentation?
-treatment?
-proliferation of blood vessels
-newborns; no, usually within first few days/months after birth
-single lesion
-they involute within a few years
Nevus Simplex
-based on appearance, also called a __ or __
-malformation of __
-commonly found where?
-appearance?
-temporary/permanent?
-similar to Nevus flammeus, which appears on the __, and is (temporary/permanent)
-salmon patch; stork bite
-capillaries
-back of neck or eyelids
-pink/red macule
-temporary
-cheek; permanent
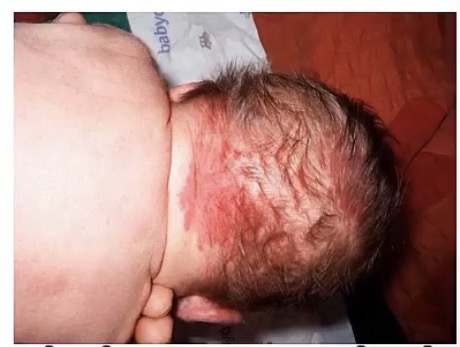
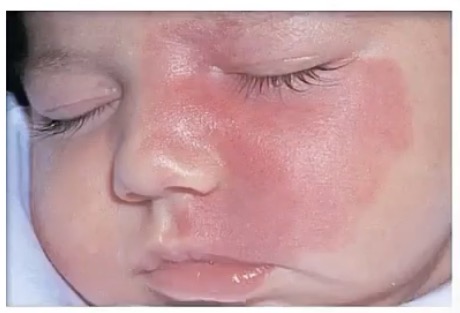
Nevus flammeus
-also called __
-malformation of __
-appearance?
-temporary/permanent?
-highly associated with what congenital syndrome?
-similar to Nevus simplex, which appears on the __, and is (temporary/permanent)
-port wine stain
-capillaries
-pink/red patch
-permanent, grows with child
-sturge-weber syndrome
-back of neck or eyelids; temporary
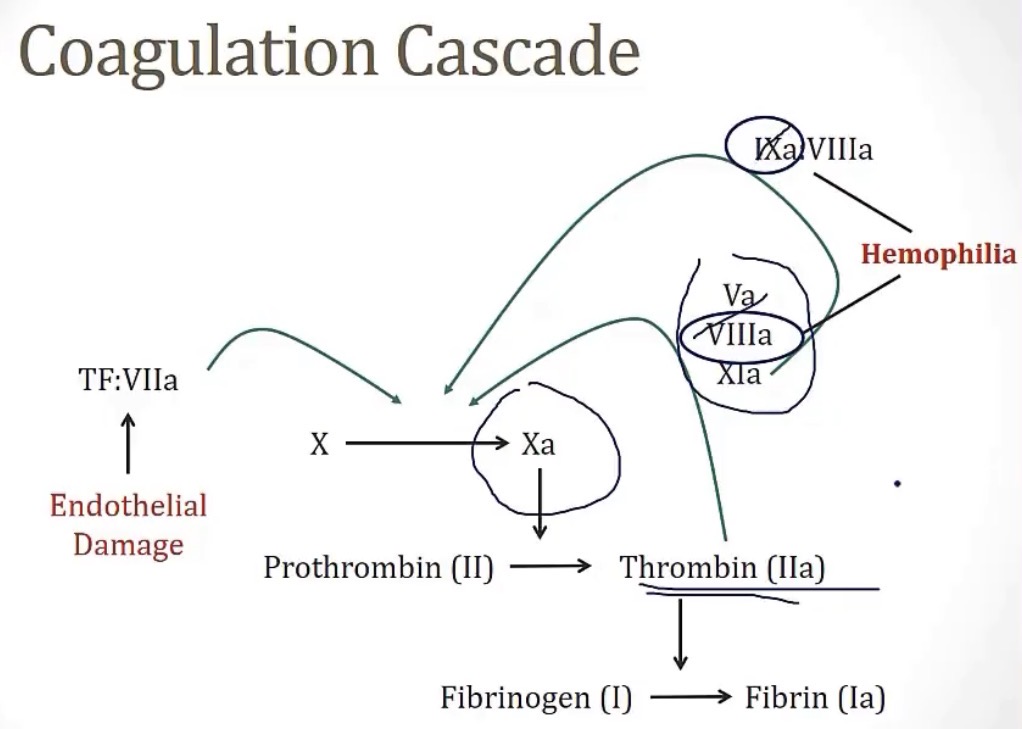
Coagulation factors are what type of enzyme?
-serine proteases
The coagulation factor that both intrinsic and extrinsic pathways converge on, is coagulation factor __
-X(10)
Tissue factor, also called thromboplastic, is expressed in __ cells
-so this circulates in the blood ONLY when…
-meaning this is considered the __
-this goes on to actuate factor __
-sub-endothelial
-there’s damage to the vascular endothelium
-major activator of coagulation
-VII
Thrombin(coagulation factor __), once activated, has a huge __ factor, and can go on to activate factors __
-one important note here is Hemophilia, which depending on type, has an inability to form either coagulation factors __ or __, meaning they lack the ability to __
-II; amplification; 5, 8, and 11
-8 or 9; amplify the coagulation cascade
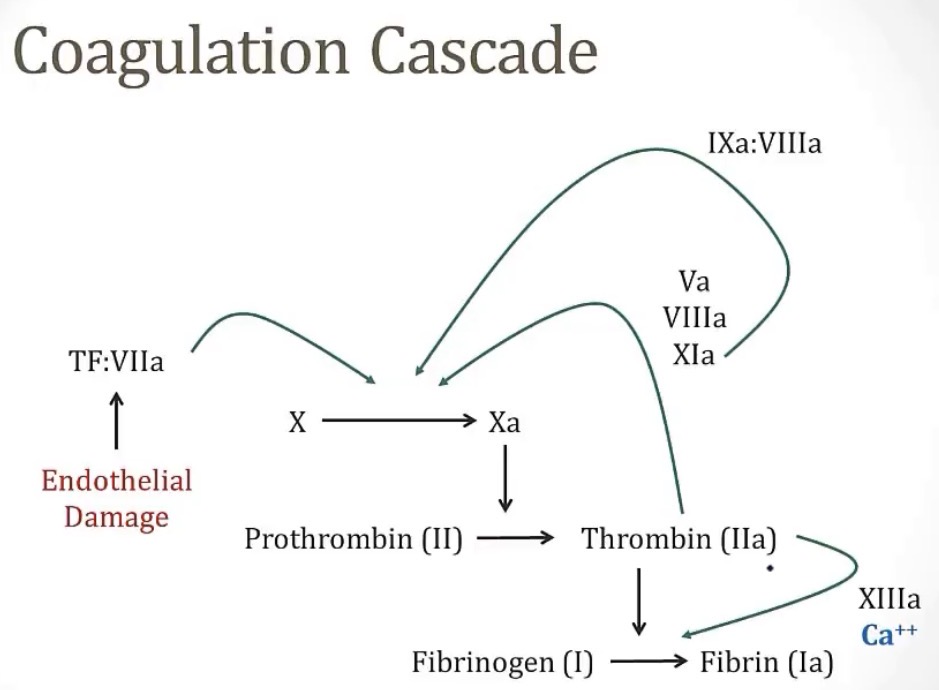
Coagulation factor VIII circulates in the blood bound to __ until there is vascular injury, where it is activated and separated by __
vWF; thrombin
Coagulation factor XIII has a job of __ to __
-it requires the co-factor __
-activated by __
-crosslinking fibrin; stabilize the fibrin plug
-calcium
-thrombin
Bradykinin
-is a __
-produced by __, which is important in relation to coagulation
-degraded by __(why you can develop angioedema from taking __)
-also degraded by __(why you can develop angioedema hereditary angioedema from __)
-vasodilator
-coagulation factor XII
-ACE; ACE inhibitors
-C1 inhibitor protein; C1 inhibitor deficiency
Prekallikrein deficiency
-individuals with this lack the ability to produce __, which will lead to an increased (PT/PTT)
-patients with this normally (do/don’t have) an issue with bleeding; why?
-bradykinin; PTT
-don’t have; it’s not necessary to kick off the coagulation pathway
Antithrombin III
-it’s classified as a __, meaning a __, which inhibits __
-activated by __, which is produced by endothelium, and this is the basis for drug therapy through __
-note that clotting factors are inhibited by endothelium, this is to prevent __ from forming in (healthy/unhealthy) vessels
-Serpin; serine protease inhibitor; coagulation factors
-heparan sulfate; heparin
-clots; healthy
Protein C and Protein S
-protein C is inactive, and becomes activated by __, which comes from endothelium
-this becomes __, which inhibits coagulation factors __ and __
-protein C requires a co-factor, __, which is always in its active form
-note that clotting factors are inhibited by endothelium, this is to prevent __ from forming in (healthy/unhealthy) vessels
-thrombomodulin
-APC(active protein C); 5 and 8
-protein S
-clots; healthy
TFPI(tissue factor pathway inhibitor)
-inactivates coagulation factors __ in 2 ways
-it either beings directly to __, or it binds to the complex of __, inhibiting activation
-10
-factor 10; factors 7 and 3
Plasminogen is converted into __, whose main role is to break down __
-2 main plasminogen activators are __ and __
-plasmin; fibrin
-tissue plasminogen activator(tPA) and urokinase
When fibrin is broken down, it produces 2 products, __ and __, and these values can be measured for diagnosis of clotting disorders
-measuring d-dimers can be very important in diagnosis of __, because it indicates plasmin is trying to break down __
-fibrin degradation products and d-dimers
-DVTs/PEs; fibrin clots
Elevated D dimers can be an indication of __, because D dimers present means attempted breakdown of __
-DVT/PE; fibrin clots
Warfarin inhibits __
Vitamin K deficiency usually presents with __
-vitamin K
-bleeding
ESR
-what is it?
-what is a high ESR show? Why?
-this will be stated in a question, that ESR is increased by __ in plasma, the key one being __
-the sedimentation rate of RBCs in a blood sample tube, basically seeing how quickly they fall to the bottom
-inflammatory conditions; there will be increased proteins, which are sticky and make RBCs clump together and settle faster
-acute phase reactants; fibrinogen
Brain break! Group bleeding disorders based on what they relate to

INR
-why is it?
-does it come from the PT or the PTT?
-used to assess bleeding time
-PT
Abnormal bleeding generally comes from abnormal levels of either platelets or coagulation factors.
If the issue stems from abnormal platelets, you can commonly see __
If the issue stems from abnormal coagulation factors, you can commonly see __
-petechiae
-joint or deep tissue bleeding
Hemophilia:
-2 types, __ and __
-note which type relates to which coagulation factor there is a deficiency of
-with this being based on a coagulation factor issue, patients will present with __
-prolonged (PT/PTT)?
-PT, bleeding time, and platelet count are (normal/abnormal)?
-treatment is __(specifically for hemophilia A, you can also use __)
-A and B
-A lacks factor 8, B lacks factor 9
-joint bleeding
-PTT
-normal
-injected replacement of lacking factors; desmopressin
Coagulation Factor Inhibitors
-what is it?
-what coagulation factor does it primarily target?
-treatment? Why?
-important note: it presents very similarly to hemophilia A, so to differentiate, you perform a __(generally describe)
-a coagulopathy based on antibodies inhibiting coagulation factors
-factor 8
-prednisone; it’s an immunosuppressant
-mixing study; mix blood with a control, and if the PTT becomes normal, then it’s hemophilia A, and if the PTT remains prolonged, it is this
Vitamin K deficiency
-causes a deficiency of what coagulation factors?
-elevated (PT/PTT)? Why?
-does this change INR?
-2, 7, 9, and 10
-elevated PT; factor 7 is crucial for PT pathway, and it has the shortest half-life of factors, so a deficiency is bad
-yes, it’s also elevated since PT is
Why can a blood transfusion cause a coagulopathy?
-treatment is __
-the blood may be deficient of clotting factors
-fresh frozen plasma
Why does liver disease cause a coagulopathy?
-clotting factors are synthesized in the liver
What is the name of the triad that leads to formation of a thrombus?
What are the 3 things?
-virchows triad
-endothelial damage, blood stasis, and hypercoagulability
Common hypercoagulable states…
-post-op, recent trauma, being still for along period of time, like plane flights, malignancy, pregnancy, OCPs, elevated homocysteine, nephrotic syndrome, smoking
In patients with an elevated homocysteine, they can take __ to lower levels
-folate
What are the 4 inherited thrombophilias?
Trick: 5 PACS(factor 5 Leiden mutation, Prothrombin gene mutation, Antithrombin deficiency, Protein C/S deficiency)
Factor 5 Leiden Mutation
-what is it?
-what is the reason for the above answer?
-what does this cause?
-this causes what?
-this is part of what group?
-abnormal factor 5
-point mutation
-there’s no activation by APC(activated protein C), so it remains active longer
-a hypercoagulable state
-inherited coagulopathies(trick: 5 PACS)
Prothrombin Gene Mutation
-what is it?
-what is the reason for the above answer?
-what does this cause?
-this causes what?
-this is part of what group?
-abnormal prothrombin
-point mutation
-excess prothrombin
-a hypercoagulable state
-inherited coagulopathies(trick: 5 PACS)
Antithrombin III Deficiency
-what is it?
-what is the reason for the above answer?
-this causes what?
-based on what’s deficient, what drug would these patients have a resistance to?
-this is part of what group?
-deficiency of Antithrombin
-gene mutations or from nephrotic syndrome
-a hypercoagulable state
-resistance to heparin
-inherited coagulopathies(trick: 5 PACS)
Protein C/S Deficiency
-what is it?
-this causes what?
-based on what’s deficient, what drug would cause a problem in these individuals? What kind of problem?
-this is part of what group?
-deficiency of Protein C or S
-lack of inhibition of factors 5 and 8
-warfarin; warfarin skin necrosis and thrombosis of skin tissue
-inherited coagulopathies(trick: 5 PACS)
Antiphospholipid Syndrome
-caused by __
-highly associated with what autoimmune disease?
-what 3 antiphospholipid antibodies are present in this condition? Which causes a false positive syphilis test?
-antibodies
-lupus
-anti-cardiolipin, lupus anticoagulant, and anti-beta2 glycoprotein; anti-cardiolipin

Platelets are derived from what cell type?
And their reproduction is regulated by what hormone?
-megakaryocytes
-thrombopoietin(TPO)
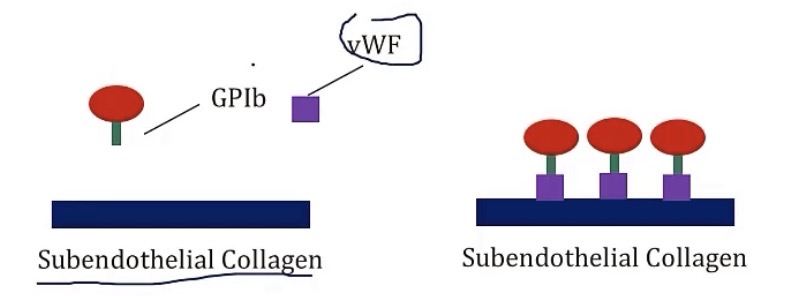
vWF(Von Willebrand Factor)
-synthesized by __
-stored in __, as well as in platelets, in their __
-circulates while bound to coagulation factor __
-it has a role of binding __ to __, by adhering to the endothelium, then binding to the __ receptor on the platelets
-megakaryocytes
-weibel-palade bodies; alpha granules
-8
-platelets to damaged endothelium; GP1b
What abundant receptor allows platelets to adhere to one another?
How?
-GP2b/3a
-by binding to either vWF or fibrinogen
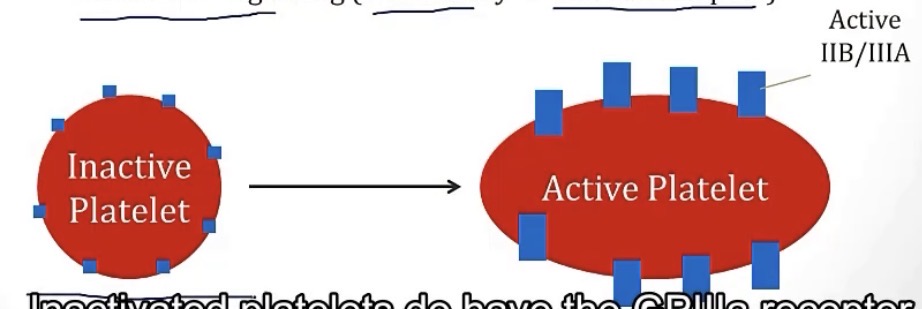
Brain break! Know what is stored in the alpha and dense granules of platelets

In Heparin-induced thrombocytopenia, antibodies form against __ complexed with heparin
-platelet factor 4
Thromboxane A2 is a medication that functions as a __, and is what’s primarily targeted/inhibited in the drug __
-platelet activator; aspirin
Glanzmann’s thrombasthenia
-what is it?
-what is the deficiency?
-diagnostic findings are __ and __
-a platelet bleeding disorder
-GP2b/3a
-isolated platelets with no clumping, and absent platelet aggregation
Bernard-Soulier syndrome
-what is it?
-what is the deficiency?
-what will this cause?
-a platelet bleeding disorder
-G1b
-platelets can’t bind to vWF, and therefore, can’t bind to the endothelium; you will also see large platelets and thrombocytopenia
Wiskott-Aldrich Syndrome
-what is it?
-what is the mutation? And what does the dysfunctional thing normally do?
-what is the triad seen in this?
-a platelet bleeding disorder
-of the WAS gene; T-cell cytoskeleton maintenance
-immune dysfunction, decreased platelets, and eczema(trick: WISkott-aldrich- W(without platelets), I(immune issue), and S(ecSEEma))
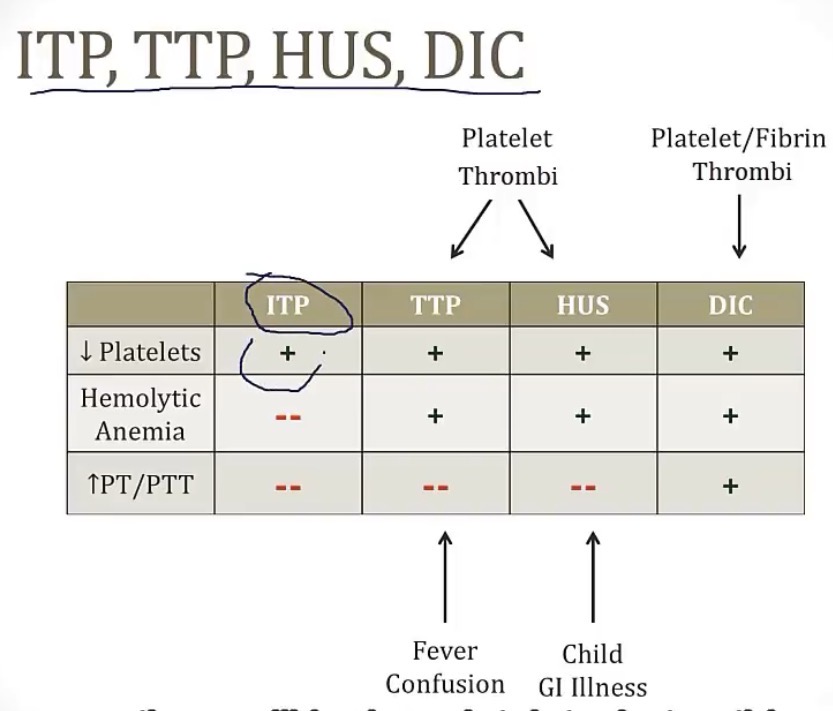
ITP(idiopathic thrombocytopenia purpura)
-what is it?
-caused by antibodies to __
-what is the source of platelet destruction, due to the antibodies?
-considering its autoimmune, treatment can include __ or __
-acquired platelet disorder
-GP2b/3a
-splenic macrophage consumption
-steroids(immunosuppressant) or IVIG(monoclonal antibodies)
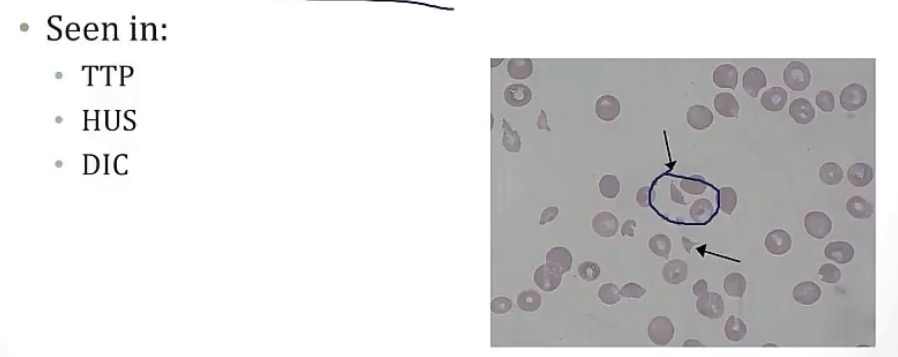
Thrombotic thrombocytopenic purpura
-disorder causing thrombus formation in __
-caused by decreased activity of __, which has the role of cleaving __
-the above generally occurs from acquired __ to ADAMTS13
-these small vessel thrombi can cause shearing of RBCs as they pass by, leading to a secondary condition called __, which has diagnostic __ cells on histology
-clinically symptoms are __, __ and __(remember, it’s a platelet issue)
-small vessels
-ADAMTS13; vWF
-autoantibodies
-MAHA; schistocytes
-neurological symptoms, fever, and petechiae
HUS(hemolytic uremic syndrome)
-what is it?
-caused by __
-presents very similarly to another acquired platelet disorder, __
-this is very commonly seen in what age group?
-commonly follows a GI infection from __, presenting with a __
-an acquired platelet bleeding disorder
-thrombi formation in small vessels
-TTP
-children
-E. coli; shiga-like toxin
Brain break! Acquired platelet disorder summary chart
Von Willebrand Disease
-what is it?
-synthesized by __ and __
-2 key roles in hemostasis…
-common symptom for women with this condition is __
-increased (PT/PTT); why?
-bleeding time (increased/decreased)?
-everything else is normal?
-diagnostic test is __(generally explain)
-treatment can include __
-deficiency of vWF
-endothelial cells and platelets
-circulates bound to clotting factor 8, and initiates adhesion of platelets(using GP1b receptor) to damaged endothelium
-menorrhagia
-PTT; remember, it’s associated with clotting factor 8
-increased
-yes
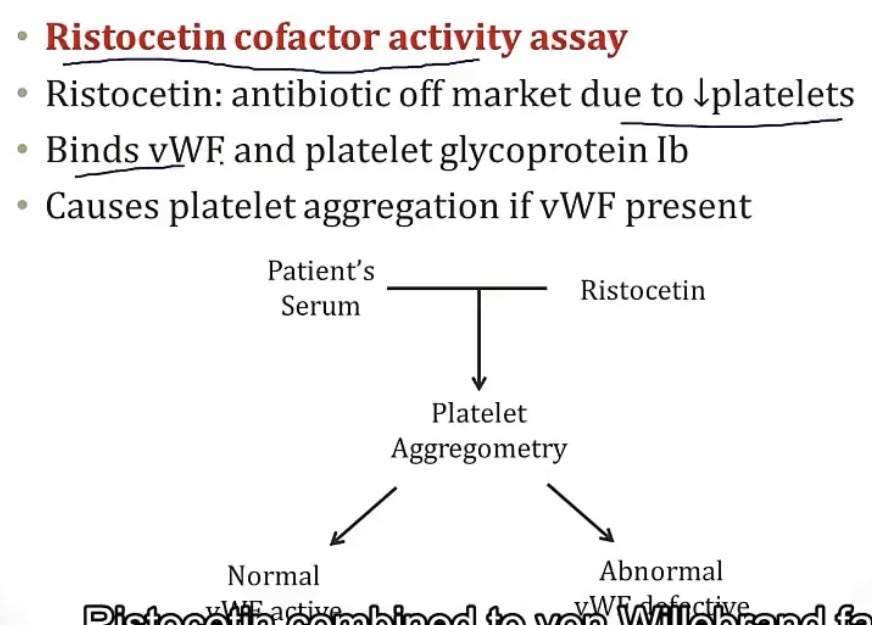
-ristocetin cofactor activity assay; makes platelets clump if there’s normal vWF
-desmopressin, aminocaproic acid, and vWF concentrate
Heyde’s Syndrome
-rare case of GI bleeding associated with __
-will have a deficiency of __; why?
-aortic stenosis
-vWF; aortic stenosis causes shearing of cells as they go through the valve, which uncoils the multimers of vWF, leaving them more exposed and subject to cleaving by ADAMTS13, reducing vWF
The role of ADAMTS13 is to cleave __
-vWF
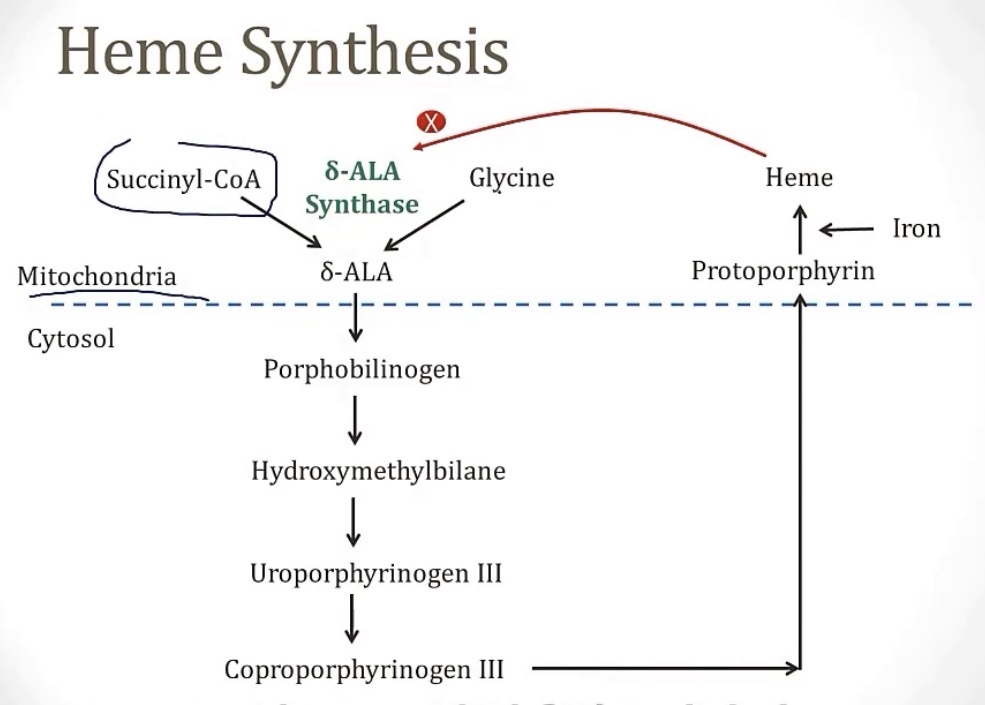
What 2 places produce heme?
-while heme primarily comes from the __, the liver produces some, which is important in synthesizing the enzyme __
-bone marrow and liver
-bone marrow; CYP450
Production of heme begins in the __ of the cell, using the 2 ingredients __ and __, and the enzyme __
-the middle steps are in the cytosol, and the final steps are back in the __
-mitochondria; succinyl CoA and glycine; delta-ALA synthase
-mitochondria
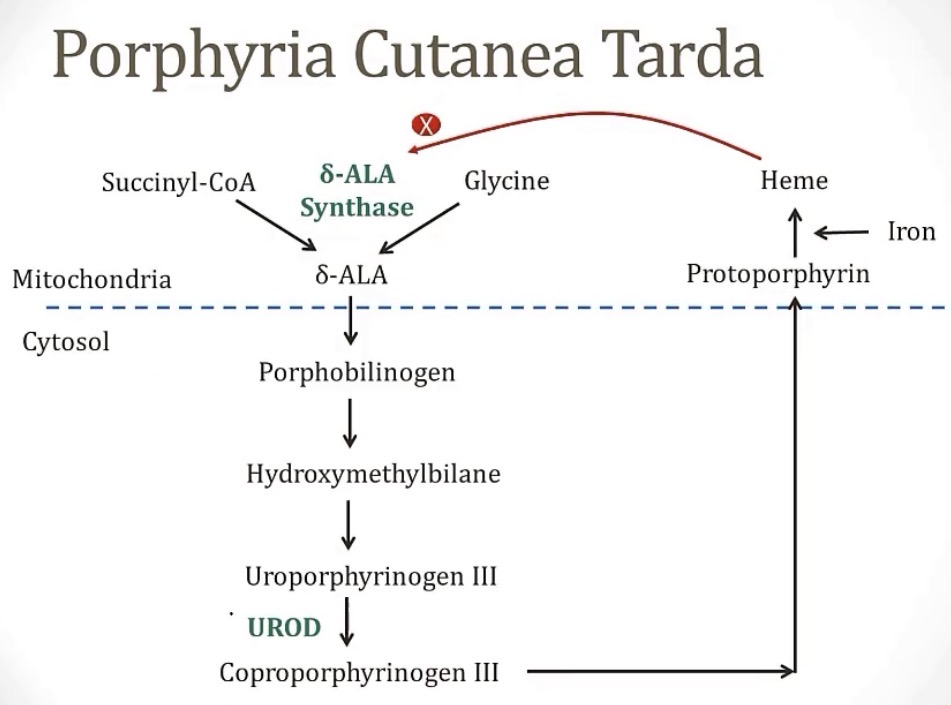
Porphyria Cutanea Tarda
-most common porphyria
-(acquired/congenital)?
-deficiency of the enzyme __, causing accumulation of __
-this accumulation is oxidized into __, which is transported to 2 places, the skin(which causes the main symptom seen, which is __), and the urine(which causes the other very common symptom of __)
-treatment is through __
-trick to remembering this: this is where the myth of vampires came from, so think about saying the name like Dracula
-acquired
-UROD; uroporphyrinogen
-uroporphyrin; skin damage on exposure to light; tea-colored urine
-phlebotomy
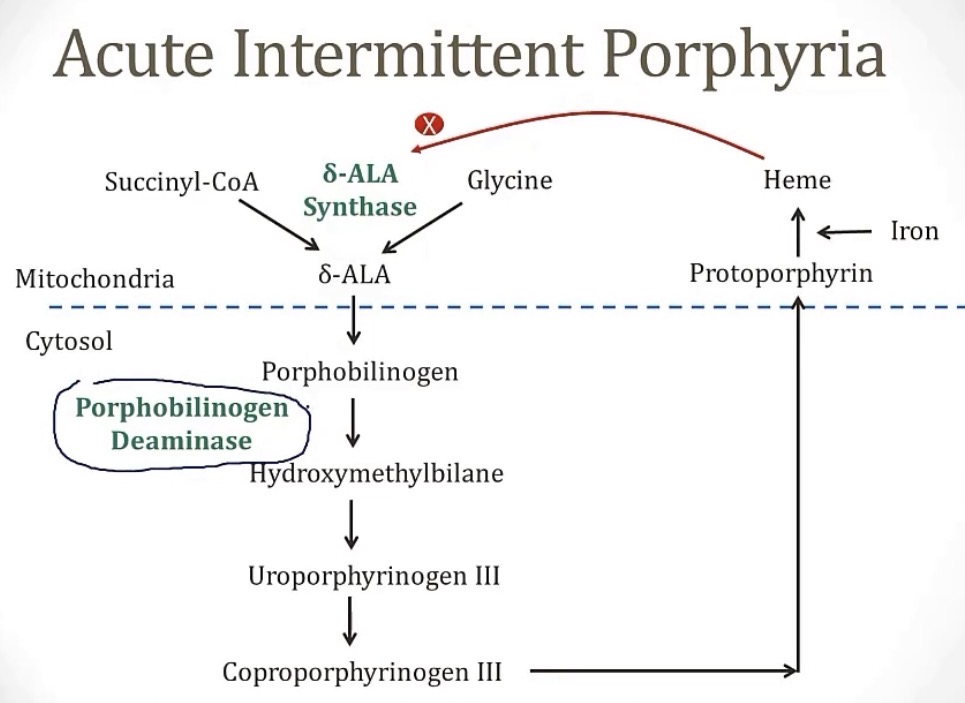
Acute Intermittent Porphyria
-what is it?
-deficiency of what enzyme?
-does have a tendency to run in families because it is __
-considering its name, these symptoms only occur in __ attacks, when the accumulation of __ rises, which is when the rate-limiting step of gene synthesis, __, is inhibited
-these “attacks” are called the 5 Ps, which are __
-what triggers the acute attack?
-best treatment?
-a porphyria condition
-PBGD
-autosomal dominant
-acute, intermittent; porphyrobinogen; delta-ALA synthase
-pain(abdominal), port wine colored urine, polyneuropathy, psychological disturbances, and precipitation by drugs
-meds that induce the CYP450 enzyme
-inhibition of heme production through glucose or hemin
What is the rate-limiting step of heme synthesis?
-delta-ALA synthase
How do we develop antibodies to blood types?
What kind of antibodies are blood antibodies?(IgM, IgG, etc.)
-from bacterial exposure from similarly structured bacteria, so they’re naturally occurring
-IgM
Brain break! ABO Blood type chart
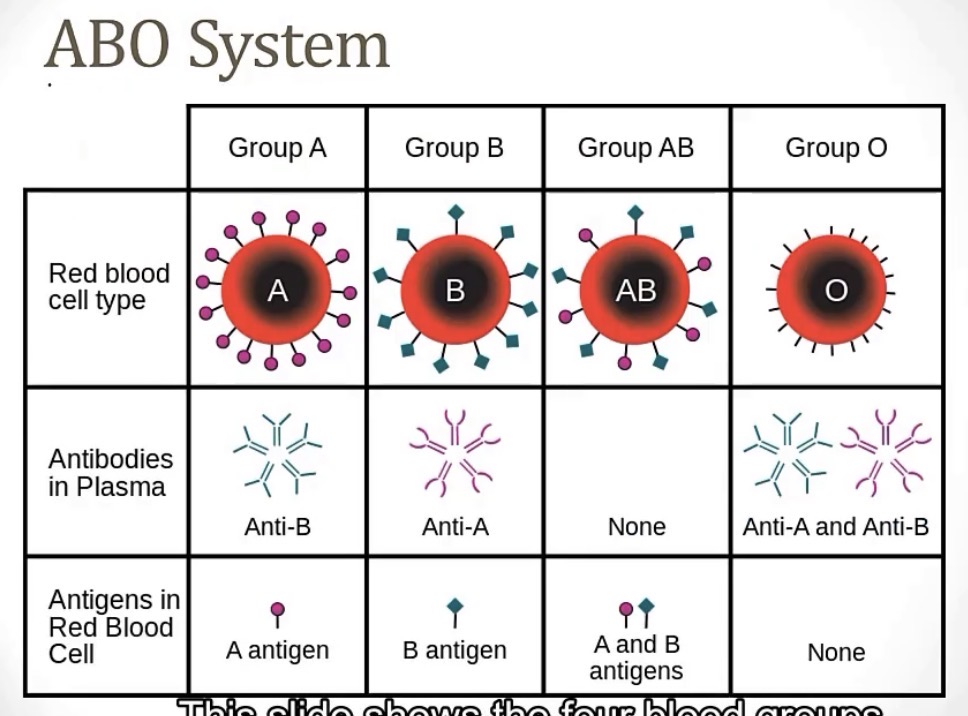
For Rh blood typing, all blood antigens are composed of __, which is different than ABO blood typing, where all the blood antigens are made up of __
Rh blood typing antibodies are of the Ig__ subtype, compared to ABO blood typing antibodies, which are of the Ig__ subtype
-transmembrane proteins; sugars
-IgG; IgM
Rh positive blood type means you (have/don’t have) the Rh D antigen
Rh negative blood type means you (have/don’t have) the Rh D antigen
Rh negative individuals CAN develop Rh antibodies against the D antigen, but only if __, such as if __
-have
-don’t have
-they’re exposed to Rh positive; a Rh negative mom has an Rh negative baby
Newborn Hemolytic Disease
-generally explain
-can this occur during a first pregnancy? Why not?
-worst complication of this is __(generally explain)
-prevention of this? How would you know a mother’s Rh status, with a direct or indirect Coombs test?
-a mother is Rh negative and has an Rh positive bay, so the mother’s Rh antibodies cross the placental barrier and attack the baby
-possibly; the first pregnancy gives the mother the antibodies, the second pregnancy is when the mother’s antibodies would attack, BUT, she could develop Rh antibodies from a prior blood transfusion, as well
-hydrops fetalis; massive edema everywhere, with heart failure, obstruction, hypertension, etc.
-if a mother is tested to be Rh negative, she will receive Rhogam, which gives her antibodies against the D antigen, so if the baby is Rh positive, anything that crosses will be destroyed; with an indirect Coombs test
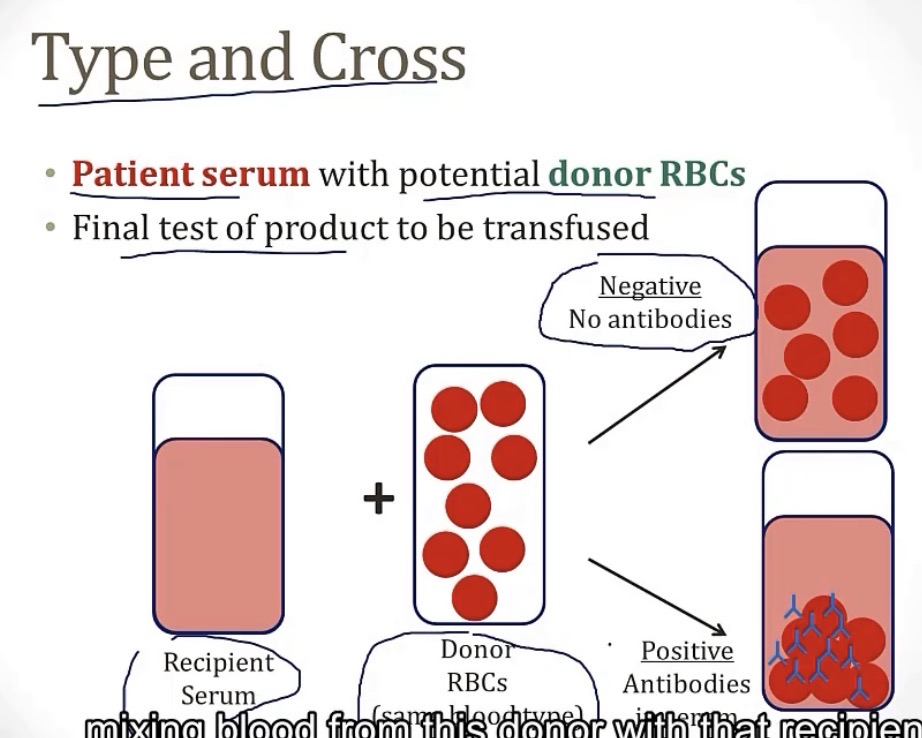
When testing for a patient’s blood type, a sample is taken and mixed with various antibodies. If you see agglutination, that means the presence of __
-antigen
What a type and cross?
-a blood type testing for any antibodies from a donor’s blood
Blood products:
•Packed RBC(generally describe)
•Platelets(generally describe)
•Fresh Frozen Plasma(FFP; generally describe)
•Cryoprecipitate(generally describe)
-RBCs with NO plasma to minimize the volume given to a patient
-for patients with low platelet count
•ONLY plasma to correct any clotting factor deficiency and normalize PT/PTT
•precipitate that forms when FFP is thawed, and has lots of fibrinogen, used for massive bleeding disorders
4 main transfusion reactions…
•AHTR(acute hemolytic transfusion reaction)
-generally describe
-what type of hypersensitivity is this?
-can lead to __
-direct Coombs test will be (positive/negative)
-usually caused by __
•Anaphylaxis
-generally describe
-what type of hypersensitivity is this?
-can classically occur in __
-treatment consists of __
•Febrile Non-Hemolytic Transfusion Reaction
-generally describe
-caused by?
-prevention?
•Tranfusion-related acute lung injury
-generally describe
-caused by?
-when recipient has antibodies against donor blood
-type II hypersensitivity
-DIC
-positive
-transfusion of wrong blood type, due to a system or clerical error
-allergic reaction
-type I hypersensitivity
-IgA deficient individuals
-stopping the transfusion and giving epinephrine
-fever and chill
-cytokines in the blood product
-some blood products undergo “leukoreduction”
-sudden onset hypoxemia during transfusion
-neutrophil activation by blood products
Normocytic anemia has an MCV between __
-80-100
Autoimmune Hemolytic Anemia(AIHA)
-based on name, generally describe
-can be warm or cold, meaning…
•Warm AIHA
-bind at body temp of __
-the antibodies are of the subtype Ig__
-symptoms include classic symptoms of anemia plus __ hemolysis, causing __)
-classic sign on histology is __
-this is most often __, but can be associated with other immune-compromised conditions
-this does have an oddly high associated with the anti-hypertensive drug __
-with this being autoimmune, treatment includes __ or __
•Cold AIHA
-bind at body temp of __
-the antibodies are of the subtype Ig__
-symptoms include __(cold stuff!)
-often associated secondary to __ by __ or __
-with this being autoimmune, treatment is similar to Warm AIHA, but most patients just __
-destruction of RBCs by autoantibodies
-what temperature the antibodies bind at
-37 Celsius
-G
-extravascular; jaundice and splenomegaly
-spherocytes
-idiopathic
-methyldopa
-steroids or immunosuppressants
-less than 30 Celsius
-M
-purple discoloration of fingers and toes
-infection; mycoplasma or EBV
-avoid the cold
Brain break! Antiglobulin tests

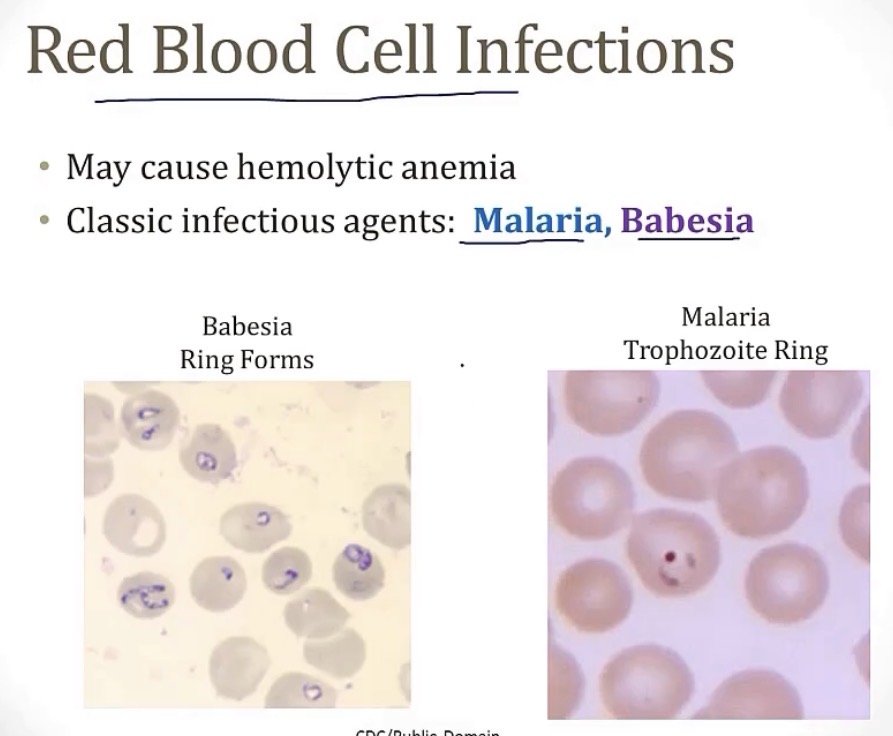
2 RBC infections that can cause a hemolytic anemia…
Hemolysis
-has a (microcytic/normocytic/macrocytic) anemia
-(elevated/lowered) LDH
-(increased/decreased) reticulocytes; why?
-(elevated/lowered) unconjugated bilirubin; will cause patient to have __ and __
-normocytic
-elevated LDH
-increased reticulocytes; hemolysis increases EPO, which produces more RBCs, and reticulocytes are just young RBCs
-elevated unconjugated bilirubin; jaundice and pigment stones(gallstones with bilirubin)
Reticulocyte count is measured in percent, and in a normal patient, it is around __
-in a patient with anemia, it is closer to __
-1-2%
-4-5%
Hemolysis can be intravascular or extravascular
-if intravascular, that means RBC destruction occurs in __, mostly from __
-if extravascular, that means RBC destruction occurs outside of __, mostly in 2 organ, the __ and __
-blood vessels; mechanical trauma
-blood vessels; liver and spleen
Hemoglobin A is made up of what 2 chains?
Hemoglobin B?
Hemoglobin F?
What test is used to determine which of these are dominant in someone?
-alpha2/beta2
-alpha2/delta2
-alpha2/gamma2
-Hgb electrophoresis
Alpha thalassemia
-__ genes code for alpha chains, and __ lead to an alpha thalassemia
-4; gene deletions

What is the carrier state of alpha thalassemia called, when you only have a deletion of ¼ alpha chains, and are asymptomatic?
-alpha thalassemia minima

Alpha thalassemia
-name for when 1/4 alpha chains are mutated?
-name for when 2/4 alpha chains are mutated?
-name for when 3/4 alpha chains are mutated?
-name for when 4/4 alpha chains are mutated?
-alpha thalassemia minima
-alpha thalassemia minor
-hemoglobin H disease
-hemoglobin Barts

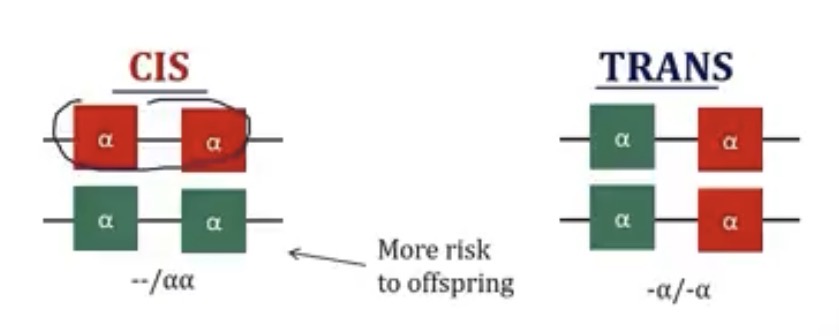
Why are there 2 variants for alpha thalassemia minor? And what are they?
Which has more risk of passing the condition on to their offspring? Why?
-it depends on if the deleted alpha chains are on the same chromosome; cis and trans
-the cis form; if they pass off the chromosome that’s deleted, the offspring already have 2/4 mutated copies of the alpha gene
In Hemoglobin H disease, where you have very little __ produced, what dominantly takes over to fill in the gap?
What is Hemoglobin H? What are its properties?
What property of RBCs will be present? And what will this lead to?
In these patients, Hemoglobin electrophoresis isn’t a reliable test, because some alpha globins are produced, so instead, __ is used
-compatible with life?
-with treatment being blood transfusions, what is a long term risk?
-alpha globin; beta globins
-the name for 4 beta chains together; 10x more affinity to O2, so it will bind O2 but not release any into the tissues
-abnormal RBC deformability; the RBCs will no longer be flexible and compliant, but instead stiff and rigid, causing an extravascular hemolysis and splenomegaly
-DNA testing
-yes
-iron overload
What is someone receiving long-term blood transfusions at risk of developing?
-iron overload
In Hemoglobin Barts, where you have no __ produced, you will see __ formed in utero
-compatible with life? What occurs?
-alpha globins; 4 gamma globin chains
-no; hydrops fetalis
Beta thalassemia
-how many genes code for beta globins? Different from alpha, which is coded by __ genes
-this is different from alpha thalassemia, because in alpha thalassemia, you see gene (deletions/mutations), but in beta thalassemia, you see (deletions/mutations)
-there are 2 types, depending on if 1 or both genes are mutated, these types are __ and __
-2; 4
-deletions; mutations
-minor and major
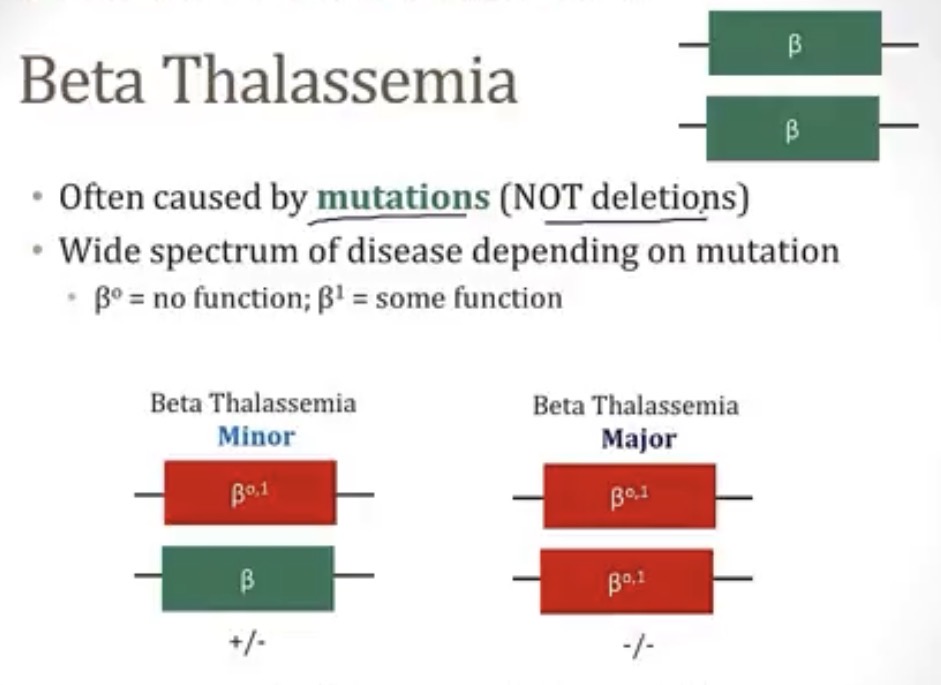
Beta thalassemia minor
-what kind of condition is it?
-what is it?
-what is making up for this lack of beta globin?
-symptoms?
-diagnosis?
-hemoglobinopathy
-condition where 1/2 beta globin chains are mutated
-increased amount of alpha chains
-no
-hemoglobin electrophoresis
Beta thalassemia major
-what kind of condition is it?
-what is it?
-what occurs?
-symptoms? If so, what?
-diagnosis?
-treatment? Risk with treatment?
-hemoglobinopathy
-condition where both beta globin chains are mutated
-ineffective erythropoiesis, where alpha chains form tetramers and cause RBC damage/abnormalities
-yes; anemia within first year of life
-hemoglobin electrophoresis
-blood transfusions; iron overload

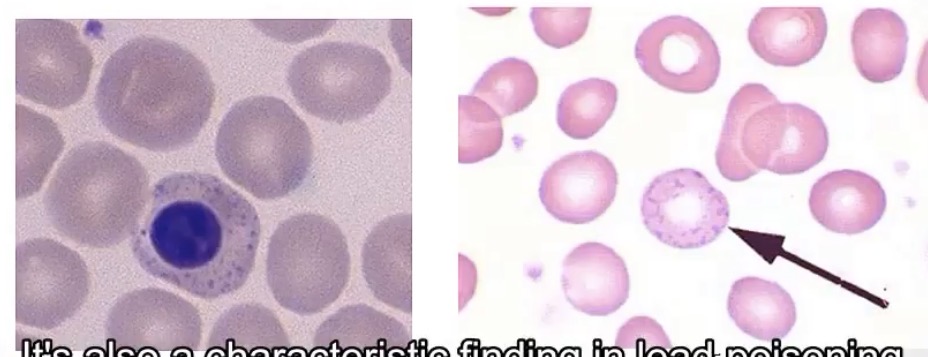
In what 2 cases will you see basophilic stippling of RBCs? What is it?
-lead toxicity and beta thalassemia major; residual RNA in red cells
Target cells
-seen any time there’s decreased __, or increased __
-the major cases you’ll see this in are __
-cell volume; cell membrane
-beta thalassemia major, iron deficiency, liver disease, and splenic dysfunction
Erythroid hyperplasia
-what is it?
-what is it classically seen in?
-it causes physical abnormalities such as __
-when there’s markedly increased EPO without normal response, so there’s a massive expansion of bone marrow
-beta thalassemia major
-chipmunk facies and crew cut appearance on x-ray skull
Extramedullary hematopoiesis
-what is it? How?
-what can this cause?
-what causes it to occur?
-what will be seen in RBCs?
-hematopoiesis outside of the bone marrow; the liver and spleen can produce RBCs in a small amount
-hepatosplenomegaly
-beta thalassemia major
-they will be nucleated
Both alpha and beta thalassemia are protective against any condition? Why?
-malaria; there’s decreased growth in RBCs of plasmodium falciparum
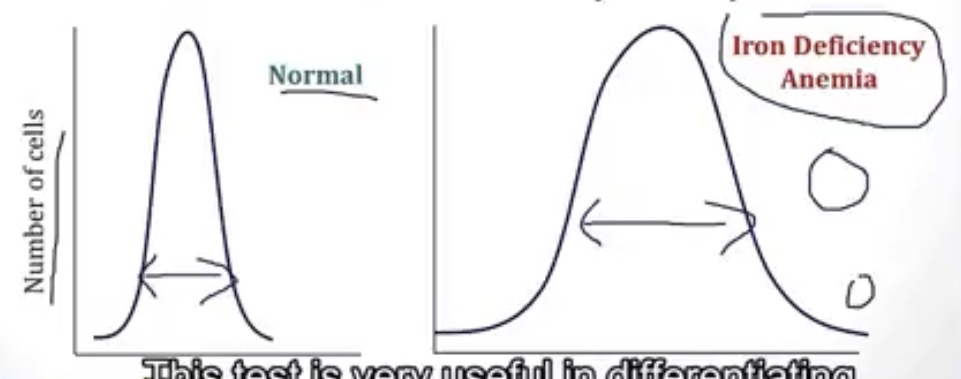
In a Red Cell Distribution Width(RDW) test, what causes a wide shape?
-iron deficiency
What is leukemia? Compared to lymphomas?
-malignant proliferation of WBCs into the blood; these are the same, but don’t appear in blood, as they’re restricted to the lymph nodes