Pharmacology Theory
1/267
Earn XP
Description and Tags
V2
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
268 Terms
Bioavailability
Rate and extent at which the API (active moiety) is absorbed from a drug and becomes available at the site of action.
Oral administration
F< 1 (variable) requires a larger dose compared to IV.
Ex. B-blockers (5mg) and Metoprolol (50mg)
Factors affecting bioavailability (F)
- Lipid partition effect (Large P means the drug is highly soluble in lipid and cross membranes easily)
- Blood flow at site of administration (high bf = high F)
- Type of administration
- Metabolism of drug (Weakly acidic drugs are ionized at alkaline pH and vice versa)
What is the primary site of ABSORPTION for most oral drugs?
Small intestine
First pass effect
Oral administration (low F value)
What is the bioavailability of rectal administration compared to oral?
Rectal administration has a LOWER bioavailability than oral (given when oral is not possible)
Parenteral administration
Delivering drugs or nutrition directly into the body, bypassing the digestive system (gastrointestinal tract).
- Via injection or infusion, completely bypassing the gastrointestinal tract (GI)
- It offers rapid action, higher bioavailability, and is ideal for, unconscious or, or uncooperative patients
- IM and SC = passive diffusion.
To increase absorption
Drug dissolved in solution, add a vasodilator
Mucosal absorption is ONLY useful when
Rapid onset is required.
Mucosal absorption
Has a limited surface area
Stomach absorption
Accumulation of weak base drugs that are then poorly absorbed due to ionization.
Large intestine absorption
- Rectum site of administration
- Reduced first pass metabolism
- Erratic and incomplete absorption
- Useful in: seizures, unconscious animals and emergencies. Ex. Rectal diazepam
Oral route is NOT advised in what animals?
Ruminants.
- Long retention in forestomachs
- Slow and unpredictable absorption
- Exception: young behave like monogastrics
Absorption from the respiratory tract
- Highly vascularized → high absorption
- Volatile anesthetics are absorbed rapidly across the
alveolar-capillary barrier
- Inhalation anesthesia
Most TOPICAL drugs have a LOCAL effect
To increase absorptive effect: massage (helps with increasing blood flow), heat, hydration
Drug molecules are INACTIVE when...
they are bound to plasma molecules (can not pass different barriers/cannot be distributed)
Only the free drug is...
ACTIVE and UNBOUND and can pass different barriers.
- It can leave the circulation, distribute in tissues, produce effects, subjected to metabolism and elimination.
- As free drug leaves circulation, more drugs unbind to restore equilibrium.
In plasma the fraction of bound and free drug molecules...
exists in equilibrium.
Acidic drugs bind to...
Albumin
Basic drugs bind to...
a-1 acid glycoprotein
Protein binding is essentially a drug reservoir
Prolong presence but delay effect
Volume of distribution
Apparent volume (not real) of fluid in which drug appears to be distributed. L/Kg BW
Drugs that are highly distributed among tissues typically require...
a higher initial dosage.
High Vd
Widely distributed in the body
Depots
- Where drug molecules can be stored and released over time.
- They prolong effects and can also prolong toxicity
P-glycoprotein
- Actively pump drugs back into the circulation
- Limits CNS drug penetration
Drug elimination
IRREVERSIBLE loss of drug from the body
Drug elimination occurs by 2 processes
METABOLISM (biotransformation) → Goal: increase H20 solubility of drug, increase renal elimination
EXCRETION (removal either as metabolite or unchanged drug)
Liver is the PRIMARY site of...
METABOLISM/Biotransformation
Drug METABOLIZING pathways PHASE I
- Highly specific and nonsynthetic reactions take place in phase I metabolism
- Aim of phase I: To make the drug more polar (more water-soluble) to facilitate phase II reactions
*Some drugs directly enter phase II metabolism.
Liver enzymes
Cytochrome P450 enzymes/CYP450 (liver enzymes responsible for phase I metabolism). Original drug is often lipophilic (fat-soluble) and CYP450 enzymes performs:
Oxidation
Reduction
Hydrolysis
After Phase I, the drug may be:
Inactivated (most common)
Still active
Activated (important for prodrugs)
PHASE II metabolism
Produce highly water soluble metabolites
Typically results in drug inactivation
Conjugation with endogenous compounds such as: glucuronic acid, glycine, sulfate, ethyl, acetyl groups.
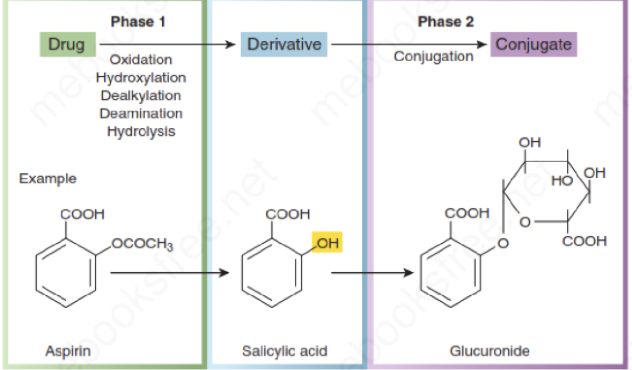
PHASE II Metabolism — CATS
Limited/ impaired phase II metabolism and conjugation
Deficient glucuronidation capacity (consequence of this is that the drugs that require a good glucuronidation capacity may accumulate → toxicity)
Paracetamol = Fatal
Metabolism — DOGS
Efficient hepatic/liver metabolism (phase I and II work well)
Exception: MDR1 mutation in some breeds → reduced drug efflux from CNS + therefore increased neurotoxicity (ex. Ivermectin)
What mutation in dogs can lead to reduced drug efflux from CNS and increased neurotoxicity?
MDR1 mutation
The main organ of ELIMINATION is the kidney. Ivermectin
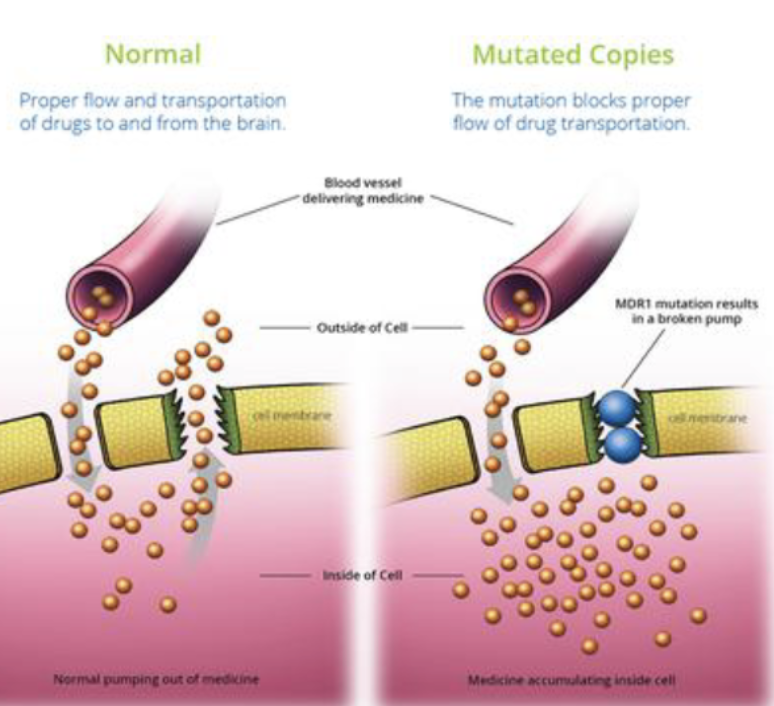
What is the main organ of ELIMINATION
Kidney
What is filtered through Glomerular filtration (passive)?
ONLY free drug fraction can be filtered
What drug fractions are secreted in the proximal tubule of the kidney?
Saturable, protein bound AND free drug fractions are secreted.
Common site for drug-drug interactions.
*Tubular secretion
Renal clearance:
Decreases with age
Reduced in renal disease
Assessed using creatinine levels
Fecal excretion:
Drugs may be excreted by bile or directly into the intestinal tract from systemic circulation.
Enterohepatic circulation:
Some drugs (especially glucuronide conjugates) are excreted into bile—> intestine—>, converted back into parent drug, reabsorbed into systemic circulation.
Consequence: prolonged drug duration, secondary plasma peaks.
Drug actions:
stimulant
depression
replacement
cytotoxic increase activity
Increase activity
functional inhibition
Endogenous substance (deficiency) can be replaced by drugs
drugs are selectively toxic
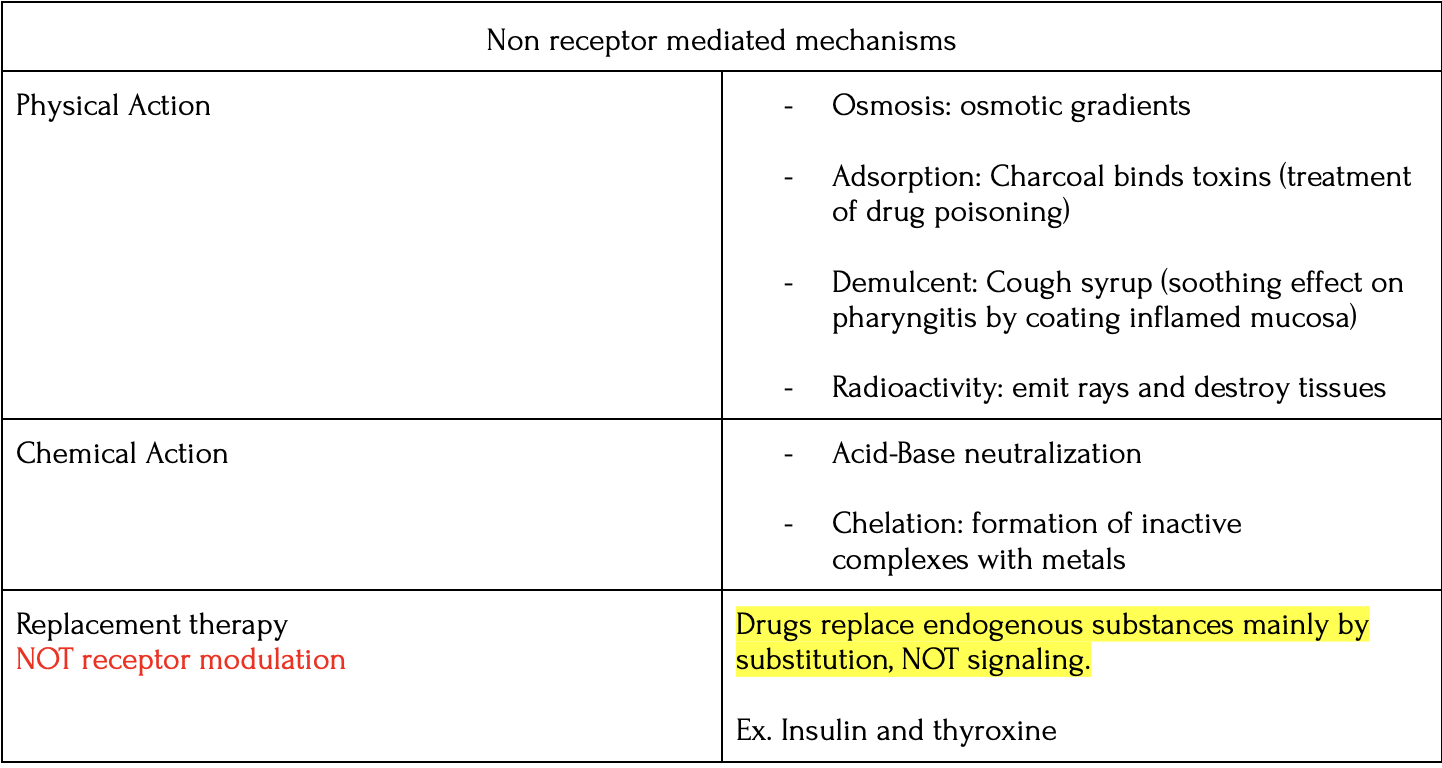
Drugs act by:
Receptor
Non-receptor: do NOT trigger transduction pathways, instead the effect is produced typically through physiochemical/ chemical properties of the drug such as shown in the table:
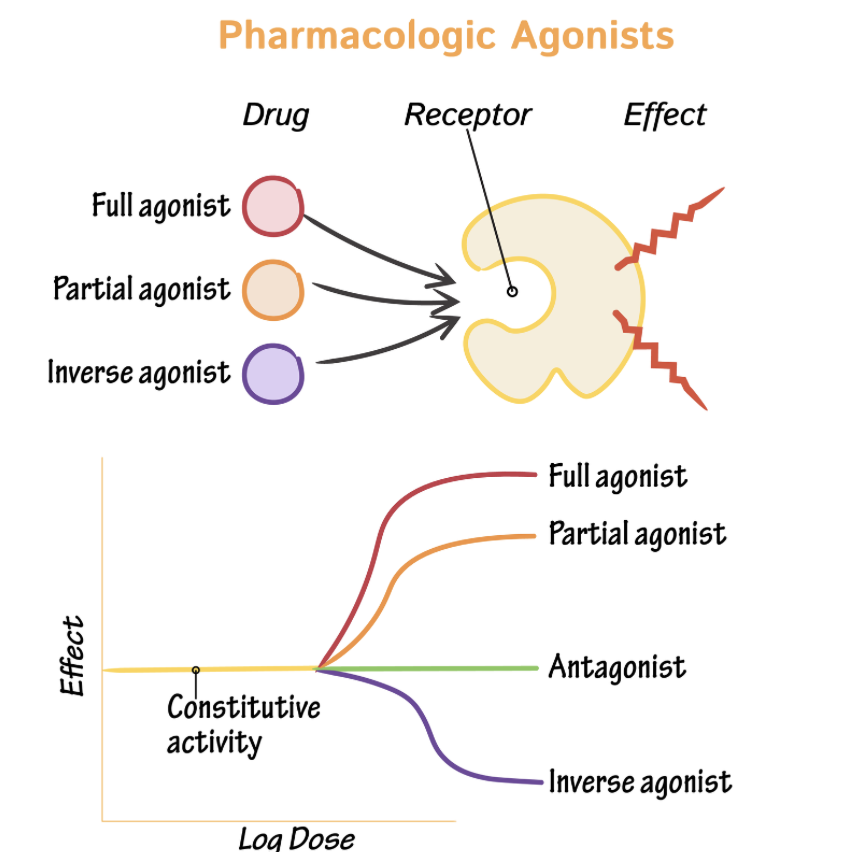
Inverse agonist
Affinity to receptor
Low/negative IA (low ability to activate receptor)
Opposite effect to agonist
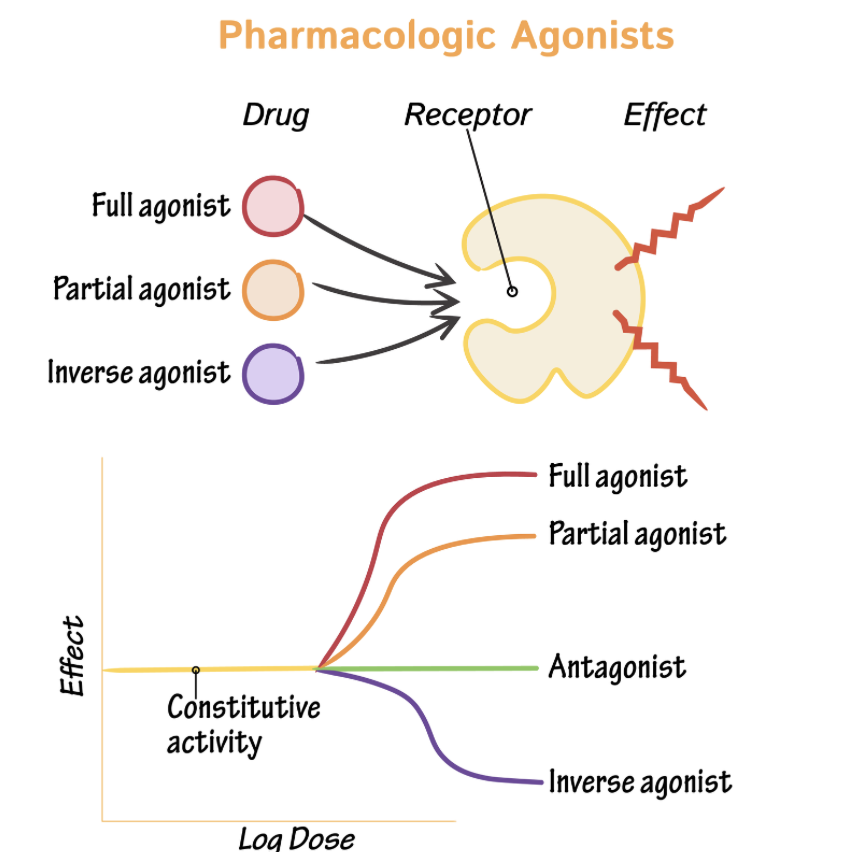
Antagonist
High affinity
NO/ZERO efficacy (IA): they have zero intrinsic activity (meaning they do not turn the receptor "on" themselves)
Block the action of an agonist or of an endogenous ligand
Their binding results in a "blocking" or "inhibitory" effect.
Antagonists cause up-regulation, which can lead to dangerous rebound effects if stopped abruptly
*Upregulation: Long-term blockage (e.g., by beta-blockers) reduces signaling, triggering cells to increase receptor density.
Agonists
Receptor desensitization → Leads to receptor down-regulation (TOLERANCE)
An agonist is a drug (or natural chemical like adrenaline) that binds to a receptor, activates it and produces a response.
Agonists: high affinity, high IA/Intrinsic Activity (Efficacy)/Activation
Mimics effect of endogenous ligand
What is an example of an intracellular receptor?
Steroid receptors
What is an example of ligand gated ion channels/ Ionotropic?
Cholinergic nicotinic receptors
hyperpolarization/depolarization
G-protein coupled receptors/ Metabotropic
Alpha and beta adrenoreceptors
Muscarinic
Smell/taste
Enzymatic receptors
Ex. Insulin
Stimulates cytosolic kinase activity
Drug potency
Amount of drug required to produce a desired response.
It is determined by affinity and intrinsic activity.
Drugs with greater potency have…
A lower dosage
Efficacy
The maximum effect of a drug (regardless of the dose).
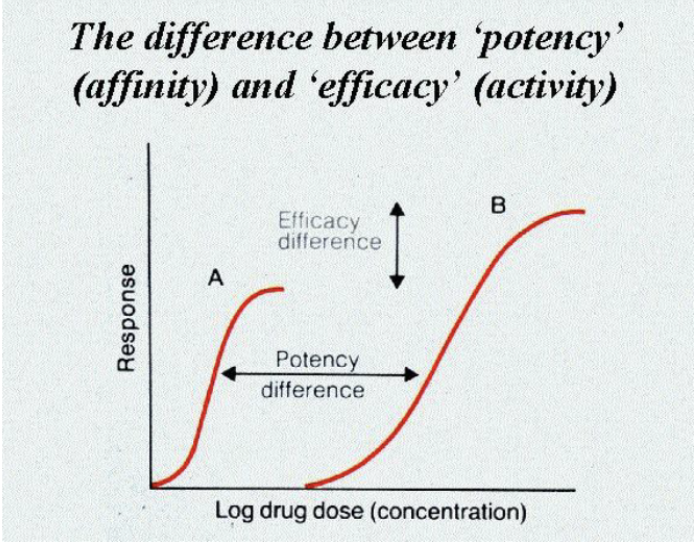
Potency
50% of Emax

Therapeutic Index (TI) should be:
TI > 1
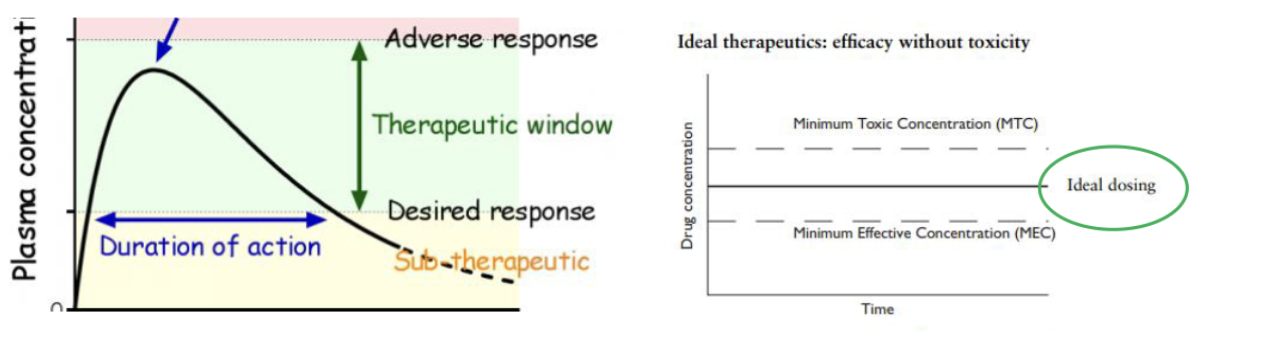
Goal of drug therapy
For drugs to be within a therapeutic window, minimizing toxicity.
Histamine
Released by immune cells, mast cells.
synthesized from the amino acid histidine
The release of histamine from immune cells can be triggered by several factors:
AG:AB (Antigen-Antibody) reactions.
Specific foods, such as crab and fish.
Bile salts
Specific drugs, which act as histamine liberators, including Morphine, d-TC (tubocurarine), dextran, and hydralazine.
Histamine acts on various receptors (H1,H2,H3,H4) throughout the body, with the highest concentrations found in…
The GI tract, lungs, and skin
H1 histamine receptors Antagonists (Antihistamines)
In the lungs, blood vessels (to induce vasodilation), heart, CNS
Located in smooth muscle (contraction), capillaries (increased permeability/vasodilation), and sensory nerve endings (pain and itching)
What are antihistamines?
Antagonists
Antihistamines (antagonists)
Antagonizes effects of histamine by competitively inhibiting H1 receptor
High specificity to H1 receptors
Used to treat allergic reactions
Anticholinergic effects
Occur due to first-generation H1-blockers having low specificity and can bind to muscarinic cholinergic receptors in addition to histamine receptors
Result from the inhibition of cholinergic transmission → dry mouth, urinary retention, sinus tachycardia, mydriasis (eyes)
H2 histamine receptors
Stomach (responsible for acid secretion)
Vasodilation
GI tract
Betahistine dihydrochloride (agonist agent)
VASODILATOR (improves blood flow to inner ear)
Histamine analogue
Agonist for receptors at CNS level
Used to cure acute peripheral vestibular syndrome (APVS)
Caution should be exercised in patients with asthma or peptic ulcers because stimulation of these receptors leads to smooth muscle contraction, which in the lungs causes bronchospasm (narrowing of the airways). Histamine analogues like Betahistine mimic the effects of histamine, they may trigger or worsen bronchoconstriction in asthmatic patients.
Any generic product that is newly released to the pharmaceutical market must show to be….
A bioequivalent to its reference product.
Bioequivalence
Shows that two products have the same efficacy (Efficacy is the maximum effect of a drug)
When two drug products show similar concentrations over time at the site of action.
Minimum Effective Concentration (MEC)
The minimum drug concentration needed to produce the desired effect.
Minimum Toxic Concentration (MTC)
The concentration at which toxic effects begin to occur
Therapeutic Window/Range
The concentration range between the MEC and MTC where the drug is effective WITHOUT being toxic
Cmax and tmax
The maximum plasma concentration
The time it takes to reach that peak
Onset Time and Duration of Action
The time required to reach MEC and the total time the drug concentration remains above the MEC
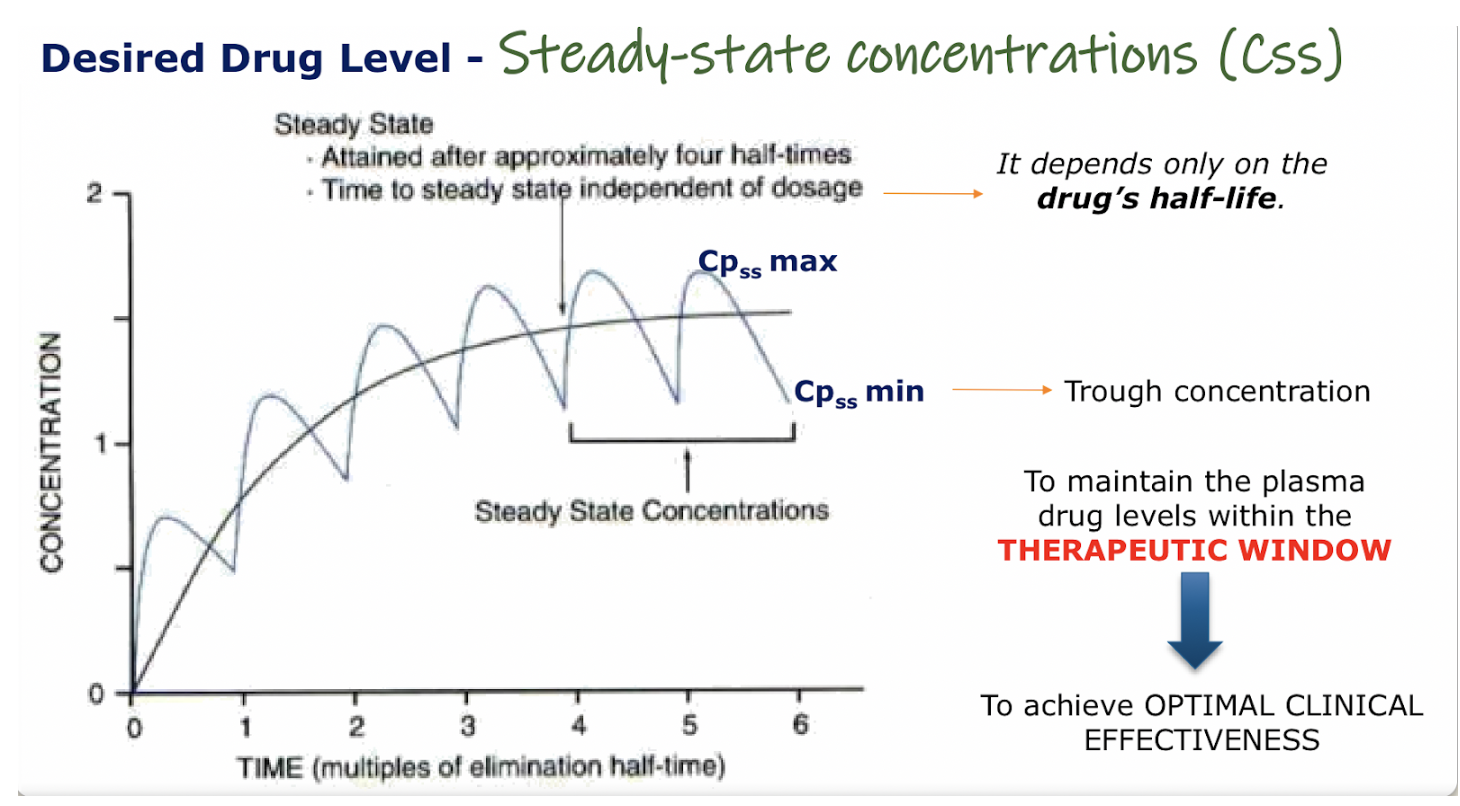
Steady state (Cpss):
Attained after approx. 4 half times
ONLY depends on drug’s half-life NOT the dosage.
Amount of drug administered during a dosing interval = Amount metabolized and eliminated.
Steady state is reached when the concentration is found between MEC and MTC (where it reaches a plato)
The two main factors adjusted in a regimen are
Dose size
Dosing interval (τ)
Loading Dose (LD)
The initial dose given to achieve target concentrations rapidly
Formula: LD=Cpss max×Vd (Note: If given orally, bioavailability (F) must be considered).
Maintenance Dose (D)
The dose required to maintain a target plasma concentration during regular administration.
Q. If a drug has a short half life but is only dosed twice daily, the concentration will spike quickly after administration and drop significantly before the next dose. What can be done to minimise this range?
To minimize this range it requires more frequent dosing.
Q. A long drug elimination halftime means the drug stays in the body for an extended period. Because the drug is cleared slowly, what would occur if one does is skipped?
The amount of drug in the system (the “steady state”) does not drop drastically when just one dose is skipped.
A long half-life acts as a “buffer” against mirror timing errors or a single missed dose.
Q. If the drug has a short half-life, what would happen to the plasma levels?
Plasma levels would drop significantly and quickly below the therapeutic range.
Q. What is the goal in a multiple-dose regimen?
To keep the drug level within a specific range; a long half-life acts as a “buffer” against mirror timing errors or a single missed dose.
To maintain plasma drug levels within the therapeutic range while minimizing toxicity.
Q. Which pharmacokinetic factor MOSTLY determines the time required to reach steady state?
Half-life of the drug
Q. A dog that requires long term treatment, the clinician wants to avoid drug accumulation and toxicity during repeated dosing. What must she do?
Extend the dosing interval to exceed the drug’s half-life
A drug with a narrow therapeutic index…
Means that the difference between a therapeutic dose and a toxic dose is very small
Q. A clinician decides to switch a patient’s medication from oral to IV in a multiple dosing regimen. Assuming 100% bioavailability with IV administration, which factor is most critical to adjust to avoid toxicity?
When switching to IV you are bypassing first pass metabolism and absorption barriers. To avoid toxicity from a sudden 100% systemic availability, THE MAINTENANCE DOSE must likely be lowered compared to oral dose.
Q. In a multiple dosage regimen, a drug that is metabolized by an enzyme subject to genetic polymorphism is found to produce unexpectedly high plasma levels in certain patients. What is the most likely explanation?
Patients are poor metabolizers due to enzyme polymorphism
What is the category relevant to veterinary medicine?
CATEGORY D (EMA)
Highest risk route for resistance
Oral group medication (feed/premixes)
local individual treatment is the LOWEST
The primary effect of a noncompetitive antagonist on an agonist is…
to reduce the maximum possible effect (Emax) of the agonist.
COX-1 ("Good Guy")
Responsible for homeostasis, including gastric mucosa protection, platelet aggregation, and renal blood flow.
COX-2 ("Bad Guy")
Inducible at the site of inflammation; responsible for pain, fever, and pathological prostaglandin production.
Therapeutic Effects of NSAIDs
NSAIDs provide analgesic, antipyretic (via PGE2 inhibition in the hypothalamus), and anti-inflammatory effects
Cats and Glucuronidation
Cats lack efficient glucuronidation. Drugs excreted as glucuronide conjugates, such as aspirin or paracetamol, have a prolonged half-life, leading to accumulation and a narrow margin of safety.
Low-dose aspirin is used for its anti-platelet effect because it…
irreversibly inhibits platelet COX-1.
Carprofen (species-specific selectivity)
COX-1 selective in humans but COX-2 selective in dogs, making it safer for canine gastrointestinal (GI) use.
most frequent side effect of NSAIDs
GI irritation
vomiting
ulceration
melena
Renal (hemodynamic acute kidney injury by inhibiting PGE2 and PGl2, which normally maintain renal blood flow through vasodilation)
Maintaining hydration and renal perfusion is essential…
for any animal on NSAIDs.
Paracetamol (Acetaminophen) Specifics:
Mechanism: It inhibits COX-3 at the CNS level and has no anti-inflammatory activity.
Feline Contraindication: Paracetamol is contraindicated in cats. They are 7 to 10 times more susceptible than dogs and can develop hemolytic anemia and hepatic necrosis.
Toxicity Mechanism: Overdose leads to the accumulation of NAPQI (a highly reactive metabolite) as glutathione stores are depleted.
Antidote: N-acetylcysteine or oral methionine is used to replenish glutathione stores.
Paracetamol (Acetaminophen) antidote:
Antidote: N-acetylcysteine or oral methionine is used to replenish glutathione stores.
Avoid NSAIDs being used with:
Glucocorticoids or other NSAIDs (greatly increases GI side effect risk).
Aminoglycosides (increases risk of nephrotoxicity).
Other high protein-binding drugs (due to competition for binding sites)