CHEM 3311 Final Exam CU Boulder
1/41
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
42 Terms
Organometallic Synthesis from Alkyl Halides
R(delta-) - M(delta+)
Rxn of alkyl halide with Mg, Li, or Na (neutral) and alkyl halide (generally Cl, Br)
Assume 1 e- transfer
Regiospecfic: C:(-) forms at C(alpha)
Stereospecific: n/a, top and bottom add'n
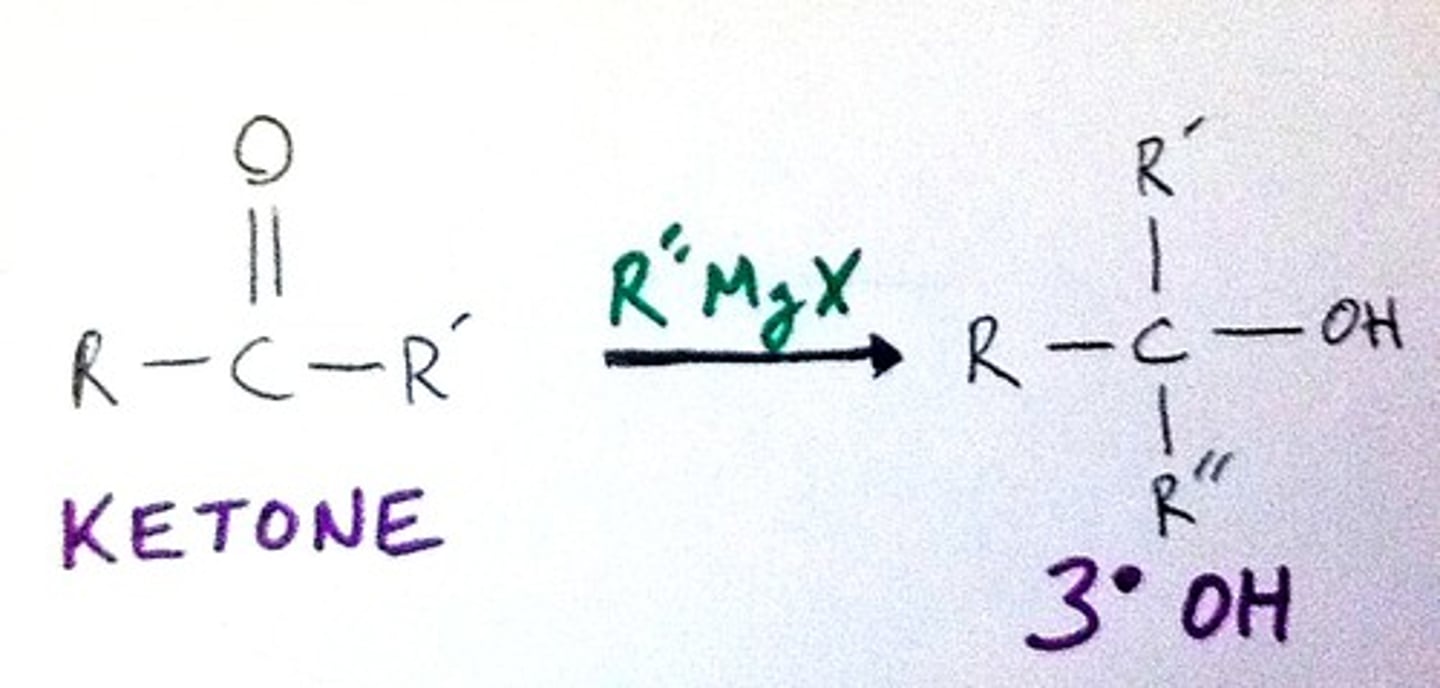
Organometallic Properties
Acts as carbanion (C:(-))
Acts as strong base if acidic H(+)s available
If carbanion is small/linear, behaves as nucleophile
Used as C chain extension reagent
Degradation of Organometallic Compound
Hydrolysis
Halogen Rxn
O2 reactions
Making/using carbenes
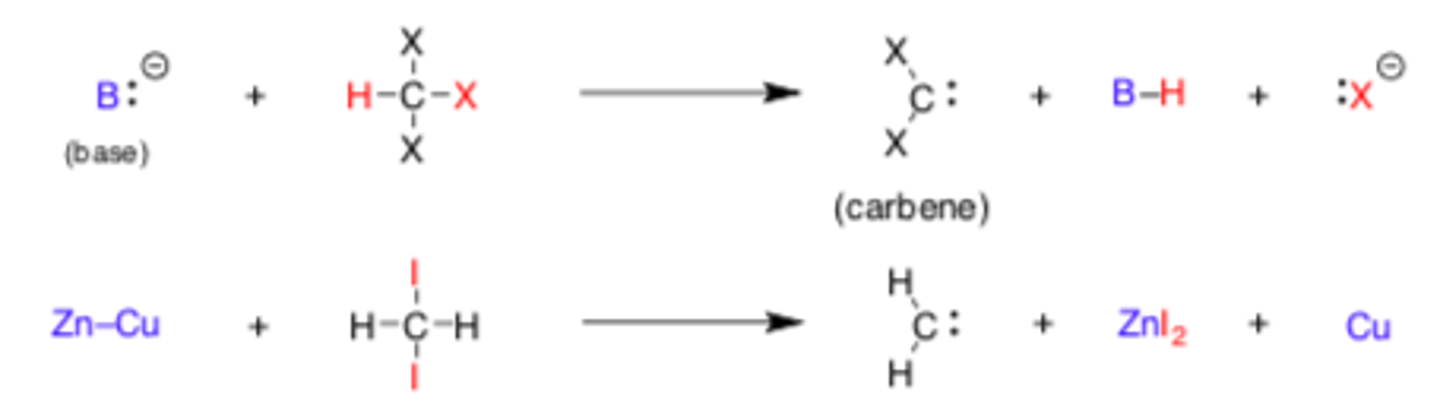
Making Carbenes from R-X derivatives
Y(2)C: + R-X derivative -> tri or di halide
If B:(+) is a strong base: di-halide, product will be trihalide+ B-H + X:(-)
If B = Zn-Cu, product will be dihalide + ZnI(2) + Cu
Hydrolysis of Organometallic
Any alcohol (-OH(-)) attacks, adds H to C(alpha) and forms salts
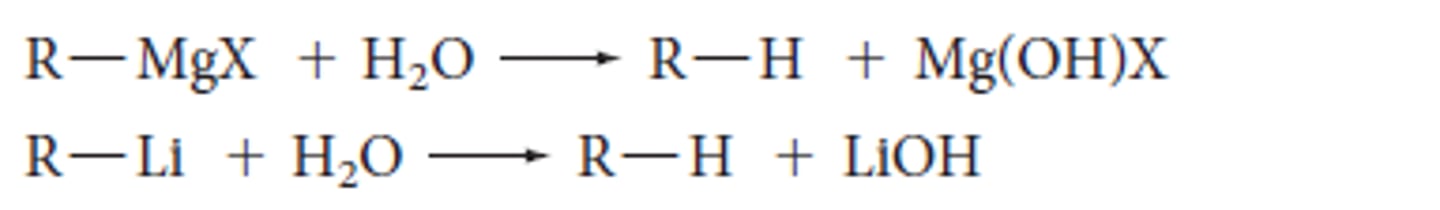
Halogen Rxn of Organometallics
X(2) with organometallis + polar (or induced polar) material via sub'n to form alkyl halide and metal compound (Li(+)-Cl(-))
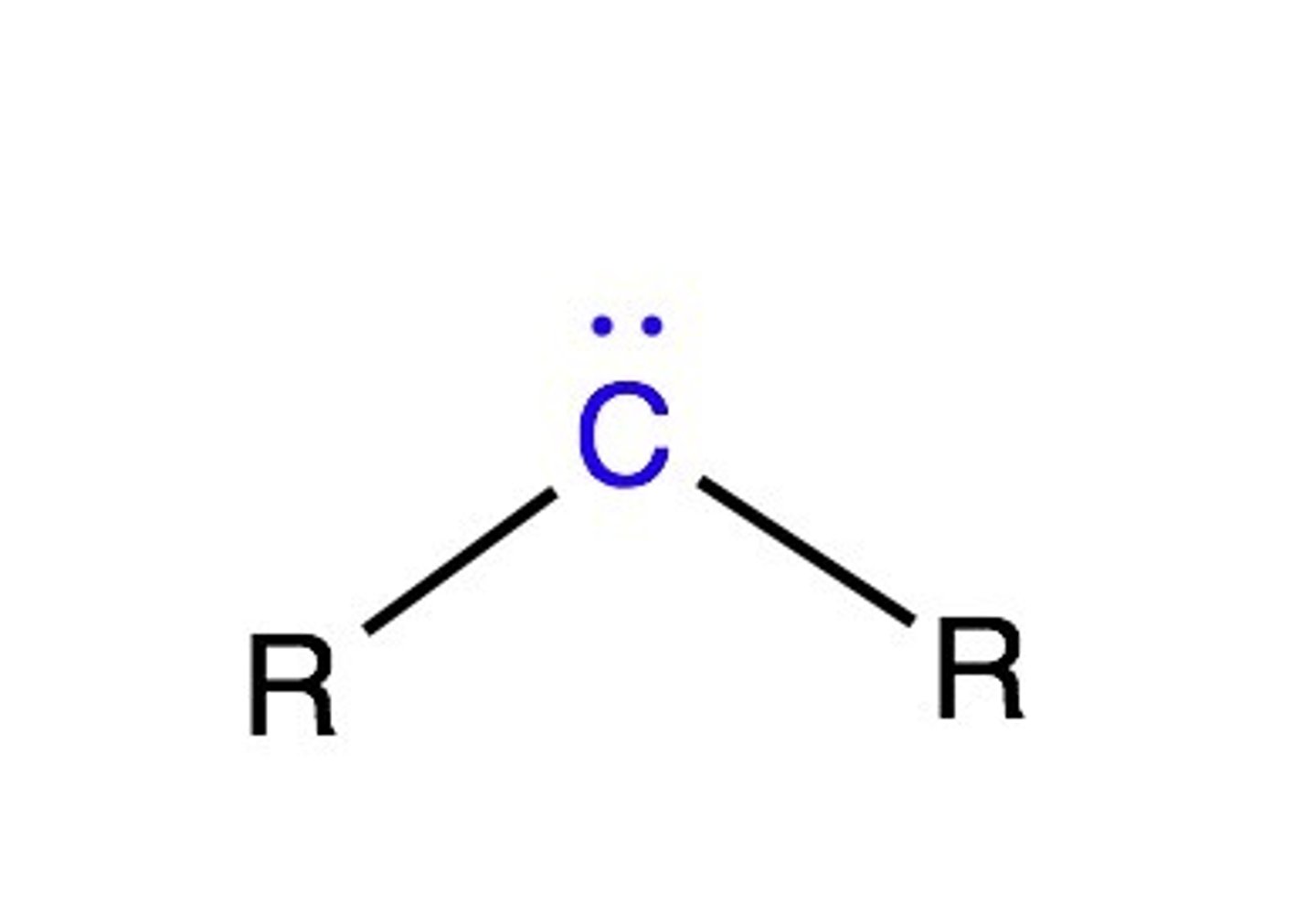
Carbene Reactant Properties
Y(2)C:
Empty p orbital -> trig planar geometry
Dual Rxn Nature: 1 group is electrophilic and nucleophilic
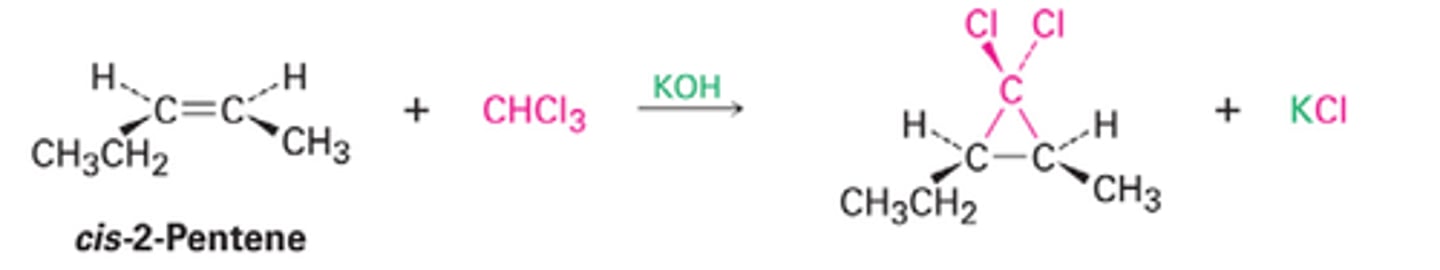
Carbene Rxn with Alkene
Add'n Rxn forms cyclopropane derivatives
No I*
Regiospecific: n/a
Stereospecific: syn add'n of Y(2), overall mix is racemic
Free radical Mono-Halogenation of Alkenes to Form Alkyl Halide
Radical substitution of R(3)C-H + X(2) -> R(3)X + H-X
Requires light/heat
Regiospecific: Br at more sub'd sites, Cl at less sub'd sites
Stereospecific: n/a, flat C radicals
Characteristics of Radical Halogenation Rxn of Alkanes to R-X
- selective for breaking C-H bonds only
- complex/multi-step Rxn with flat C.
- polyhalogenation occurs without termination of chains
- follows dihalogen trends
Dihalogenation Radical Trends
- F. Too reactive, not used
- Cl. Is reactive, faster at 1st degree C sites (less sub'd)
- Br. Less reactive, but more reactive at 3rd degree sites (more sub'd)
- I. Relatively unreactive at normal temps
Hydration of Alkenes to synth R-OH
- acid catalyzed hydration (H2SO4, HNO3 or HClO4) and H2O
- oxymercuration-reducation hydration
- hydroboration-oxidation hydration
Nucleophilic Sub'n of R-X with OH-/H2O to form R-OH
- SN2 rxn with a 1 degree R-X
- SN1 rxn of a 3rd degree R-X at low temperatures
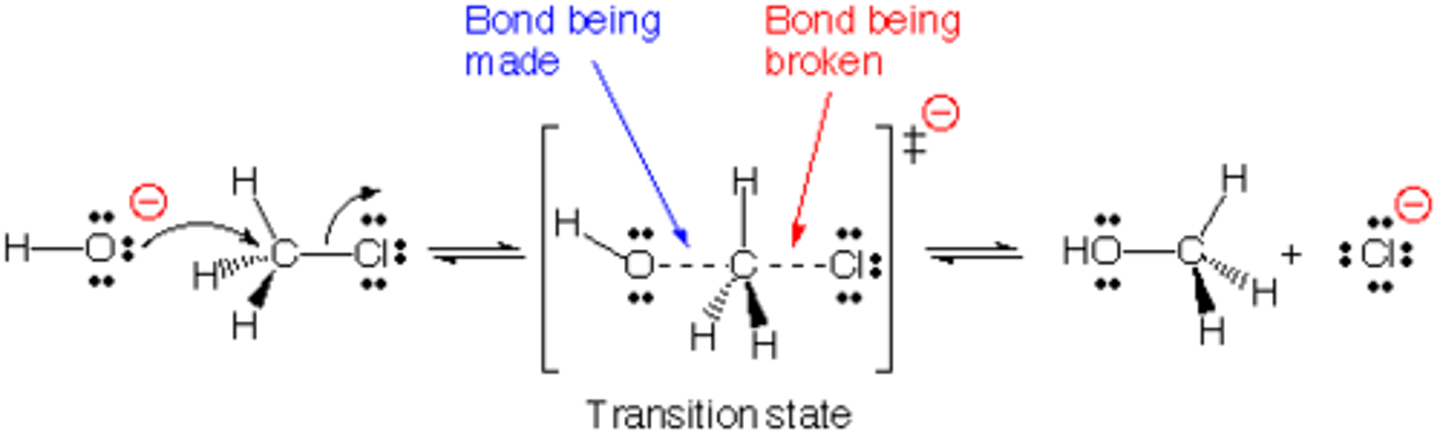
Conversion of R-OH to ether (R-OR')
- SN2 under basic conditions (Williamson ether synthesis)
- SN1 and SN2 under acidic conditions (alc dehydration)
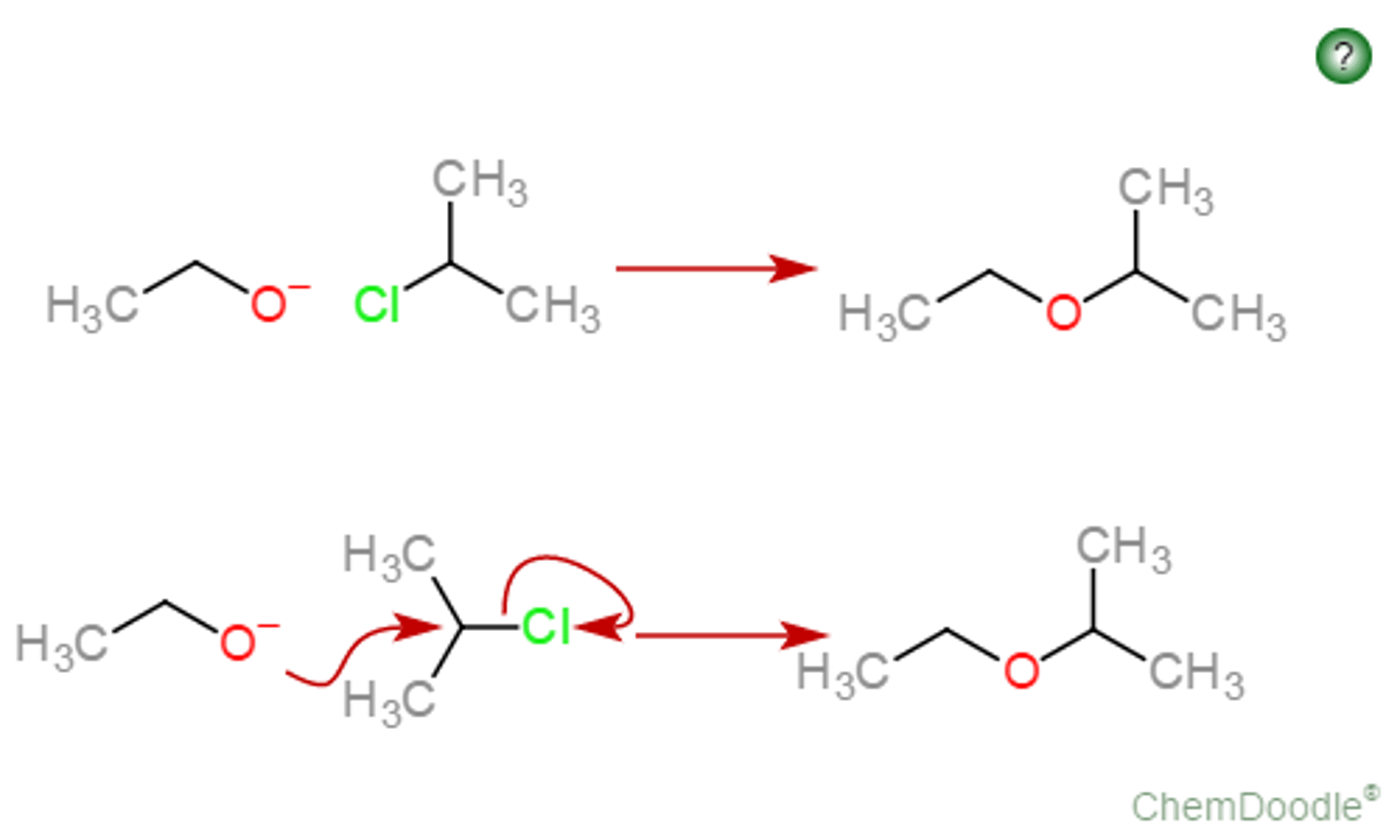
Conversion of R-OH to Alkene
acidic conditions, dehydration via elimination
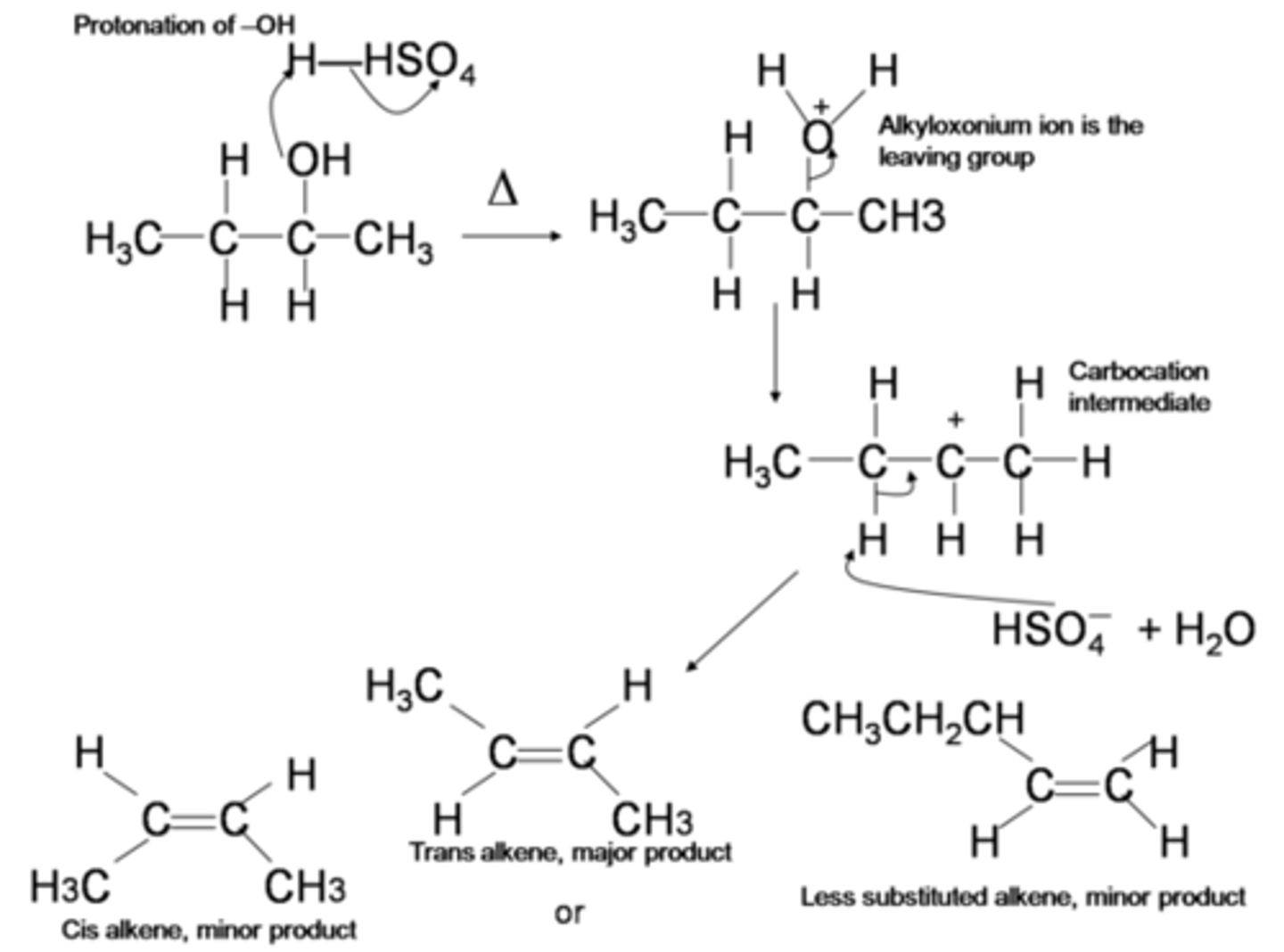
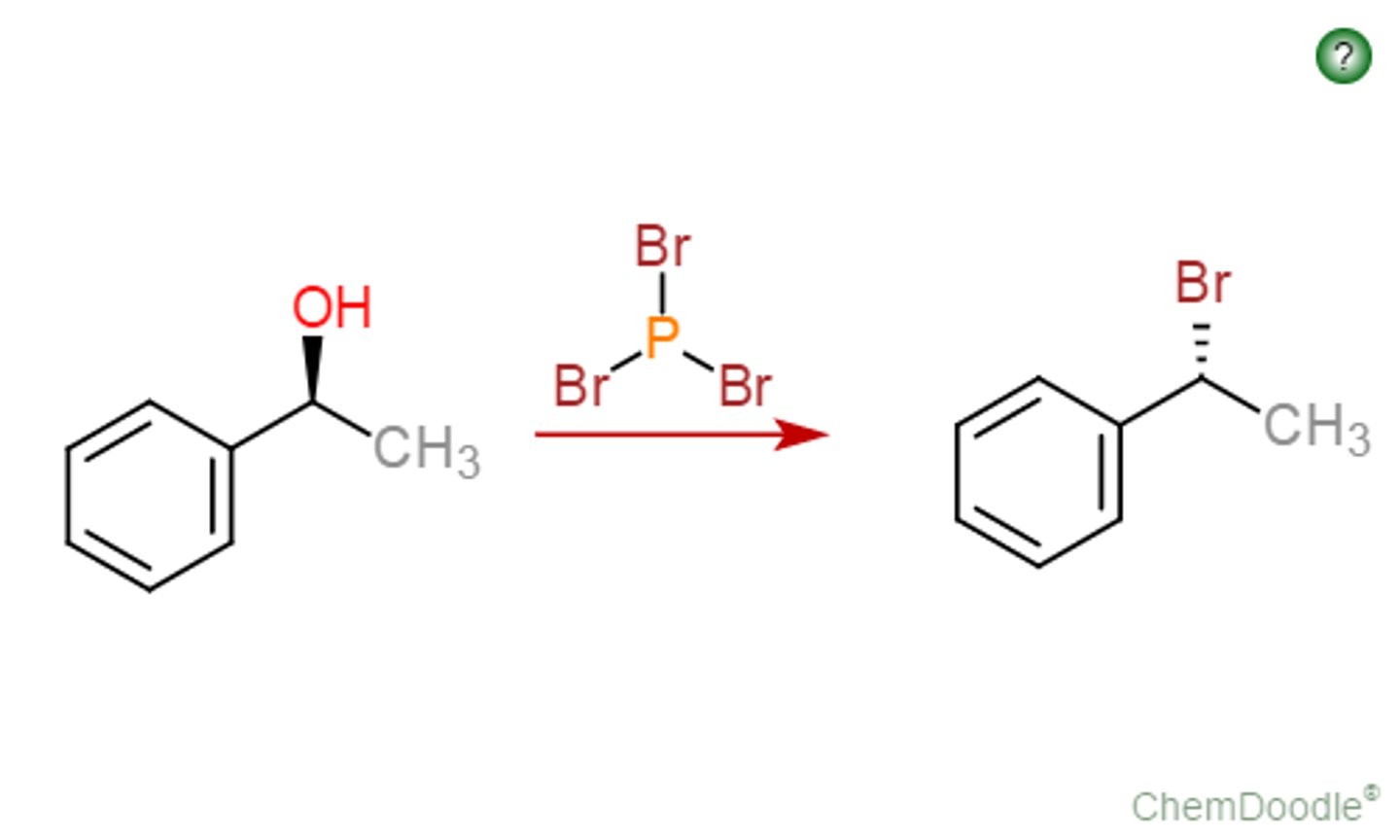
Conversion of Alcohol to R-X
sub'n of -OH for -X via:
- SN2 and SN1 rxns with H-X under acidic conditions
- SOCl2 and organic amine base (SN2) to R-Cl
- PBR3 (SN2) to R-Br
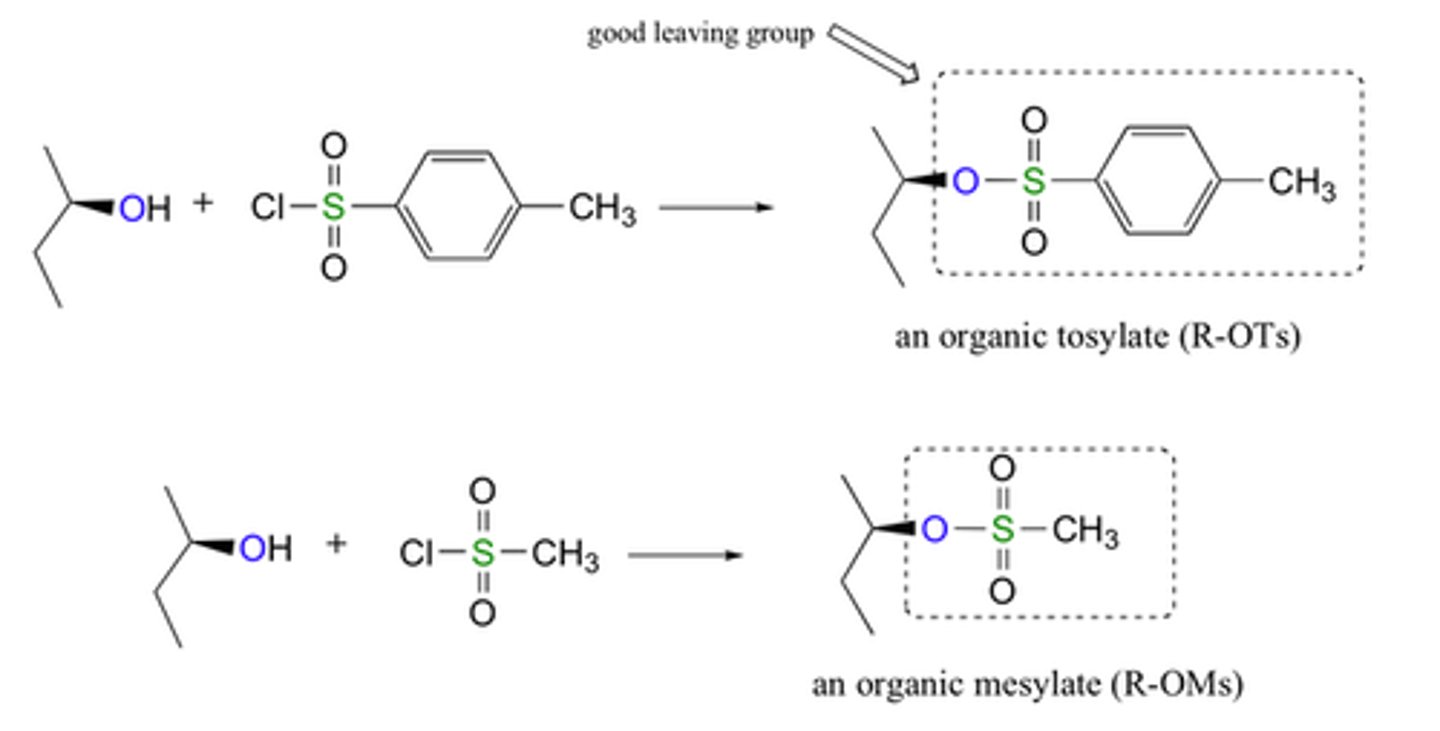
Conversion of Alcohol to Sulfonate Esters
R-OH to R-OSO2R'
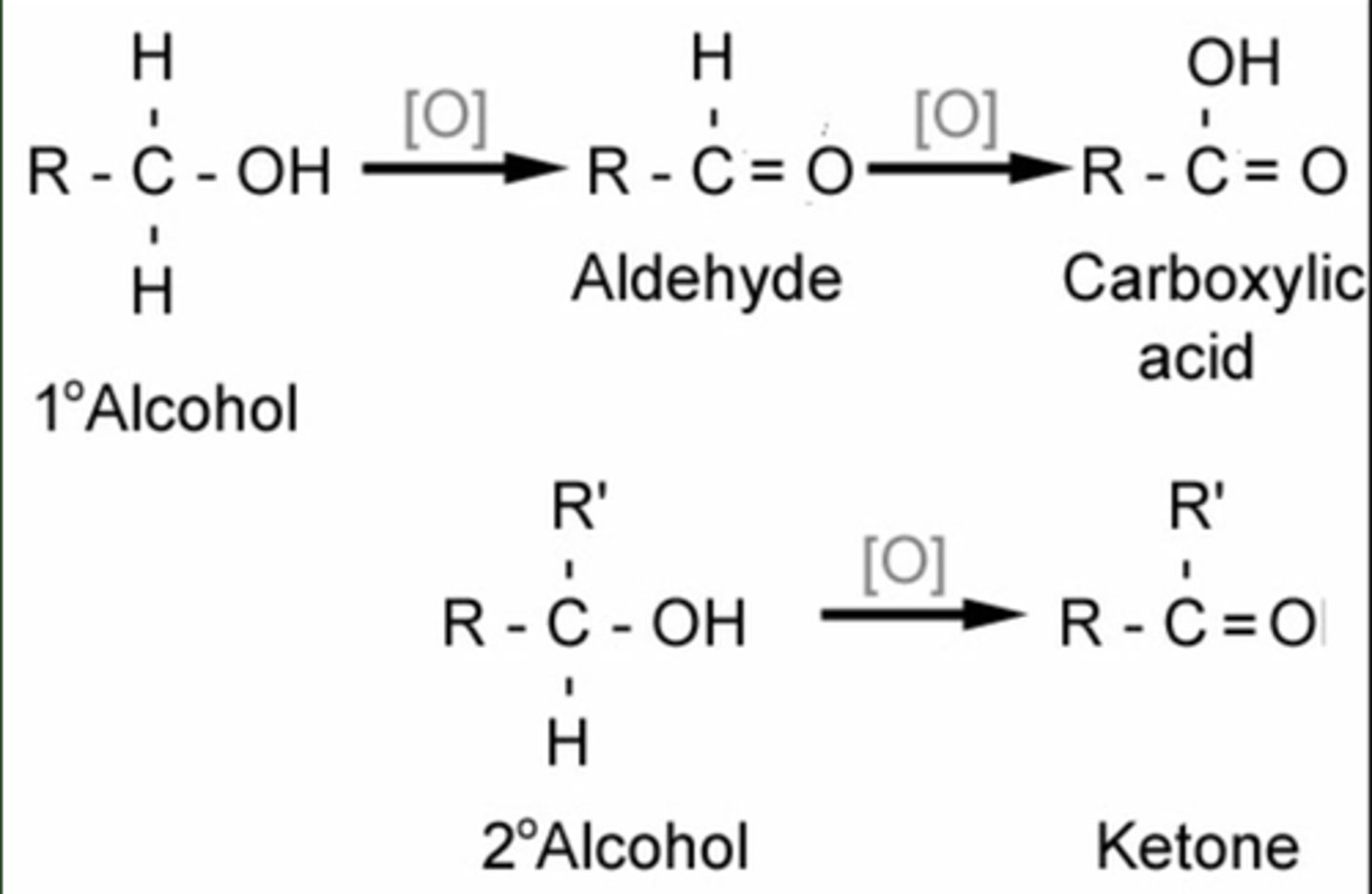
Oxidation of R-OH to Molecules Containing C=O
1 degree -> aldehyde
2nd degree -> ketone
1st degree -> carboxylic acid
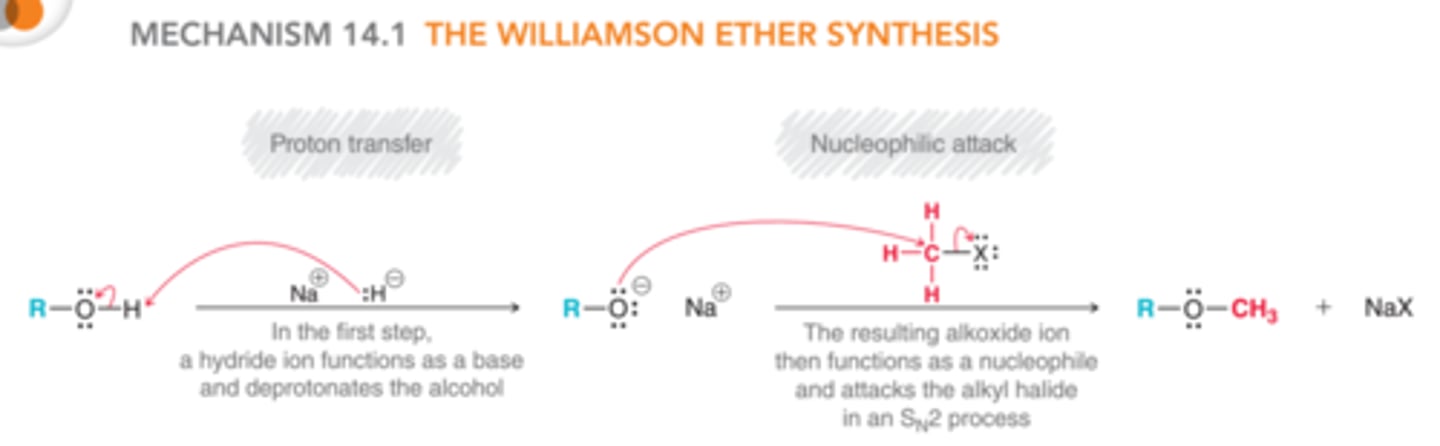
Conversion of Alcohol to Ether via SN2
(Williamson Ether Synth)
**basic conditions
1) form alkoxide anion (RO-) from ROH
2) React KO- with non-hindered R-X via SN2
- works best in 1st degree R-OH and R-X (favors SN2)
- more alpha/beta branching and strong, bulky bases has more E2 rxn products
**Regiospecific: RO- affs to C(alpha)
Stereospecific: inversion of C(alpha)
Conversion of OH to Ether (ROR) via SN2 & strongly acidic conditions
x2-1st degree ROH form a symmetrical ROR molecule
Regiospecific: ROH is NU: so adds to C(alpha)
Stereospecfic: C(alpha) inversion
Conversion of ROH to ROR via SN1 to a non-symmetrical ROR under strongly acidic conditions
3rd degree ROH with a 1st or 2nd degree ROH to form ROR'
*C+ intermediate
Regiospecific: less sub'd ROH (as NU:) adds to C(alpha)
Stereospecific: n/a, less-sub'd ROH adds to either face
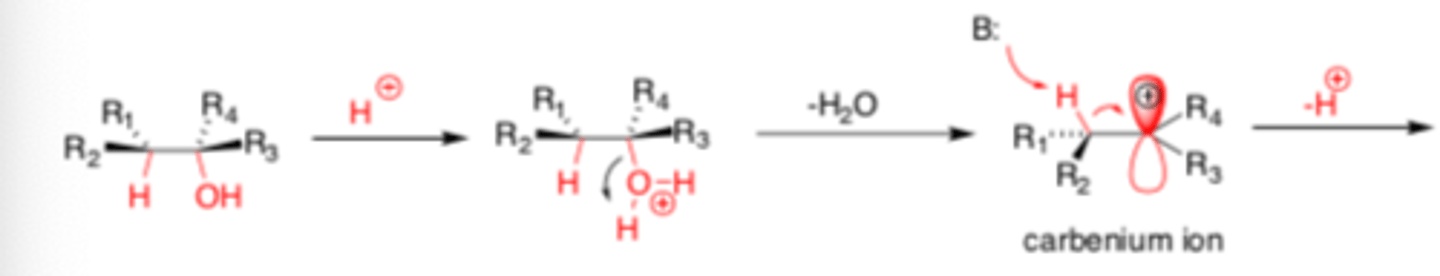
Dehydration of ROH's to form Alkenes
acid catalyzed Beta-elimination to convert -OH into good leaving group
-E1 mechanism for 3rd degree and 2nd degree ROH substrates (scrambled products)
-E2 based mechanism for 1st degree ROH substrates (stereoselective)
Zaitsev's Rule
if there is more than one Beta-alkene possible, the most substituted product will be the major product
*thermodynamic rxn control under H+-catalyzed rxn conditions
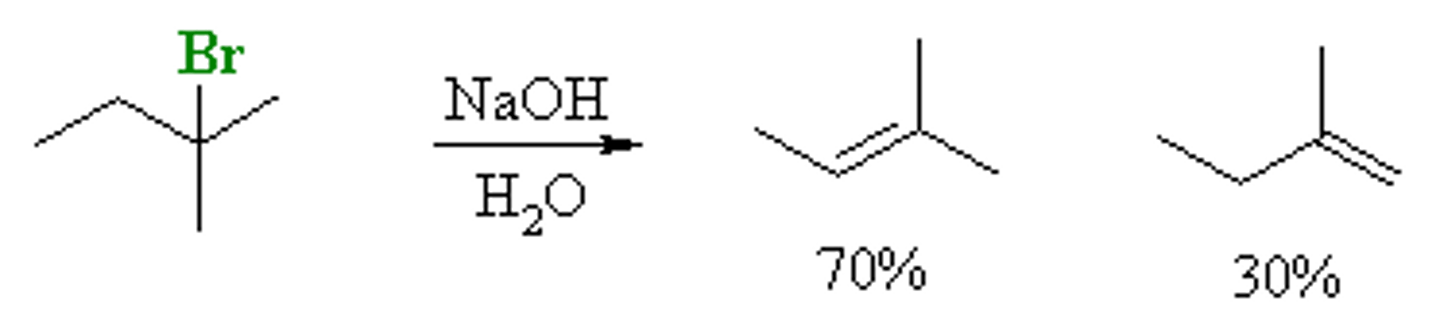
Trans-Cis Selectivity in Acid-Catalyzed ROH elimination
trans stereoisomer is major product formed (largest groups farthest apart, least steric crowding)
Exception: alkene product is cyclic molecule
E1 Elimination
Acid catalyzed: thermodymanic control, major product is most sub'd
Base catalyzed: kinetic control, major product is least sub'd
SN2 and SN1 conversion of ROH to R-X (acidic)
Acidic conditions -> brute force conversion
SN2: steroselective with C(alpha) inversion
SN1: racemic mixture
ROH to R-Cl Conversion with SOCL2
SOCL2 and organic amine base gives R-Cl
*converst R-OH to a sulfite ester (resonance stability = better leaving group)
- used with methods that react with H+
-Regiospecific: Cl- adds to C(alpha)
-Stereospecific: C(alpha) inversion
**only 1st and 2nd degree ROHs
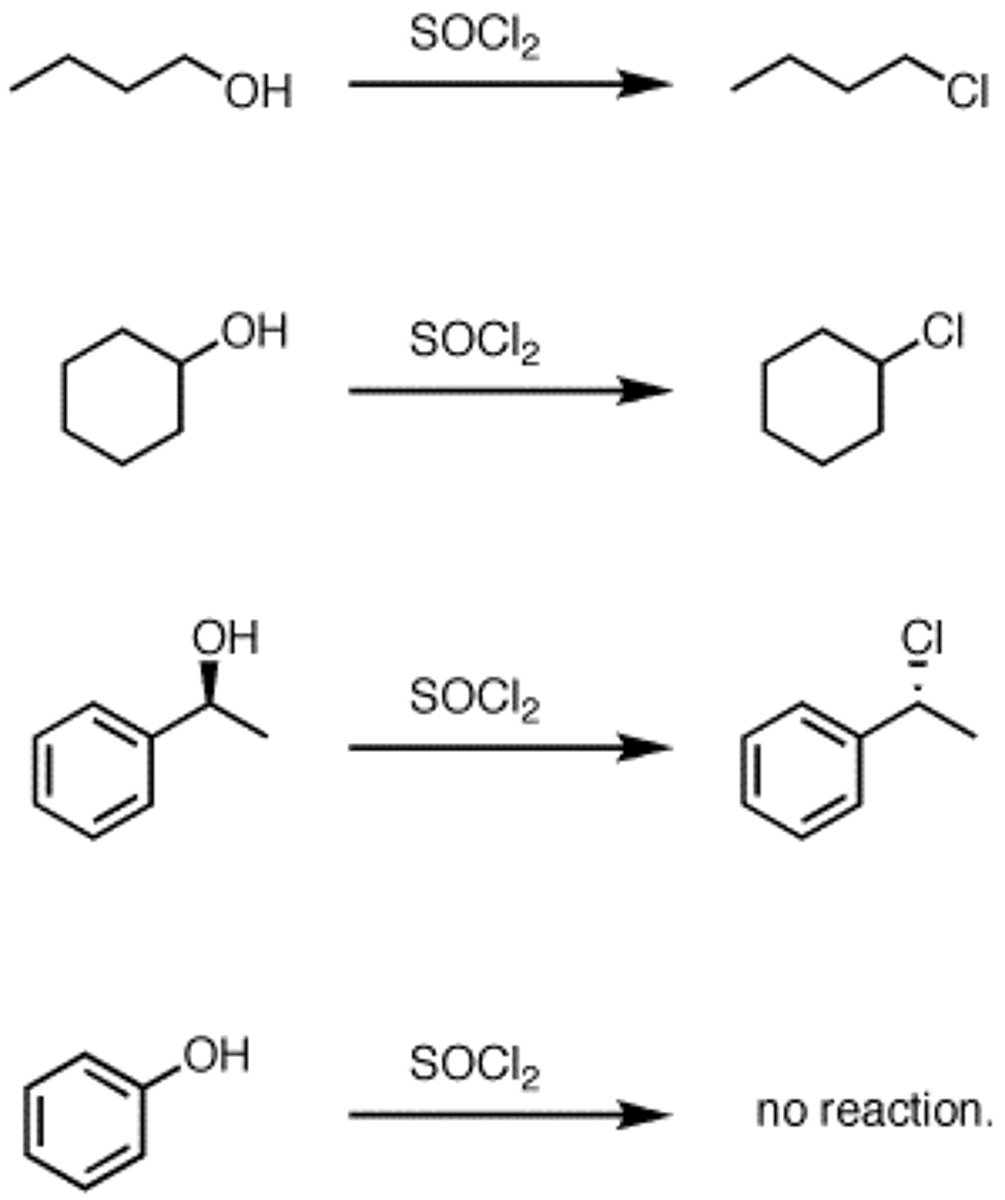
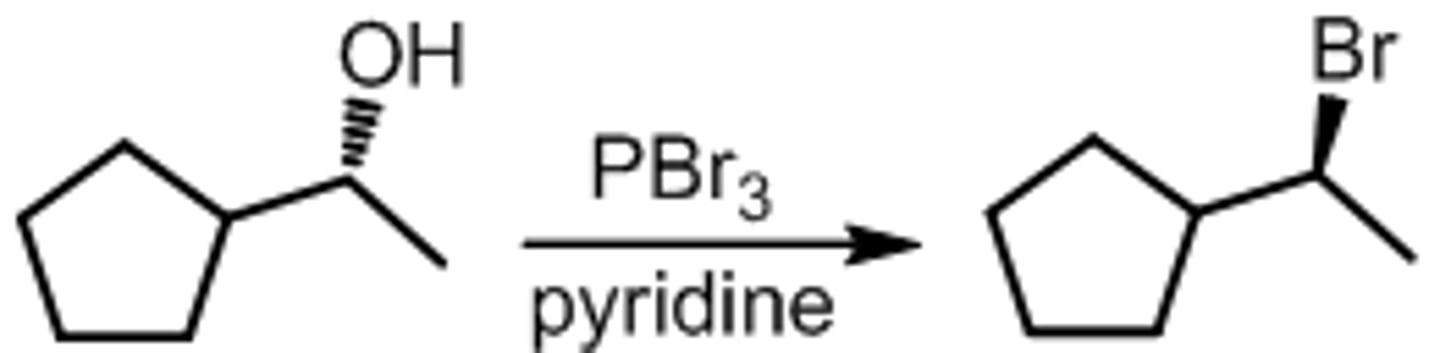
ROH to R-Br Conversion with PBr3
-OH converted to better leaving group, followed by SN2
-each -Br group replaced by RO-
- Regiospecific: Br- adds to C(alpha)
- Sterospecific: C(alpha) inversion
*1st and 2nd degree ROHs
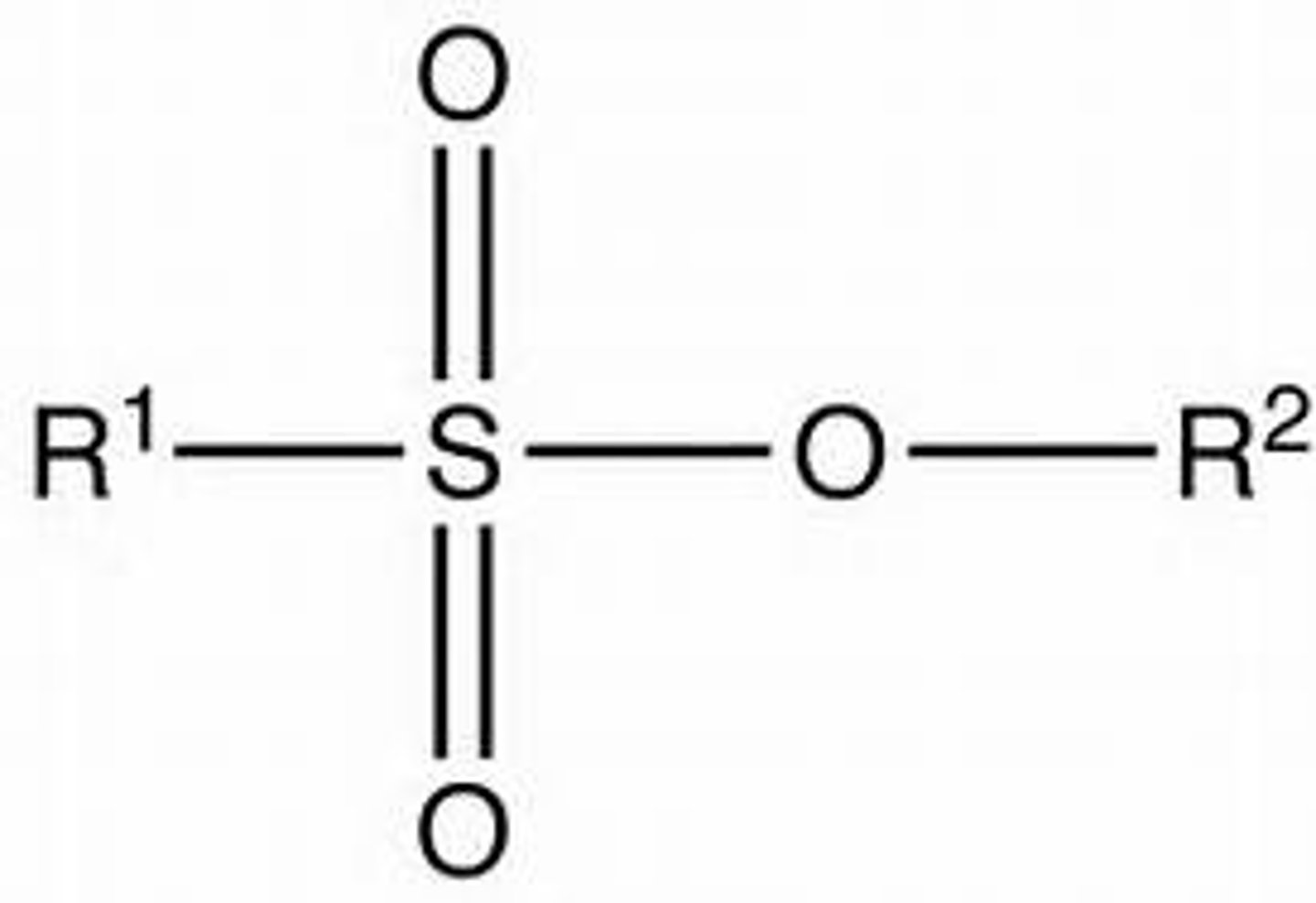
Sulfonate Ester
R-OH -> R-OSO2R' is a good leaving group (resonance stabled, not basic)
-Convert with Pyridine, CH2CL2 (results in ROSO2R' + H+ + Cl-)
Oxidation of Alcohols
1st degree ROH-> aldehyde
2nd degree ROH -> ketone
1st degree ROH -> carboxylic acid
Regioselectrive: only 1st and 2nd degree C(alpha)-OH oxidized to C(alpha)=O
Racemic mix of C=O
Oxidation
gain in # bonded O atoms, loss of bonded H atoms
Reduction
loss in # bonded O atoms, gain in bonded H atoms
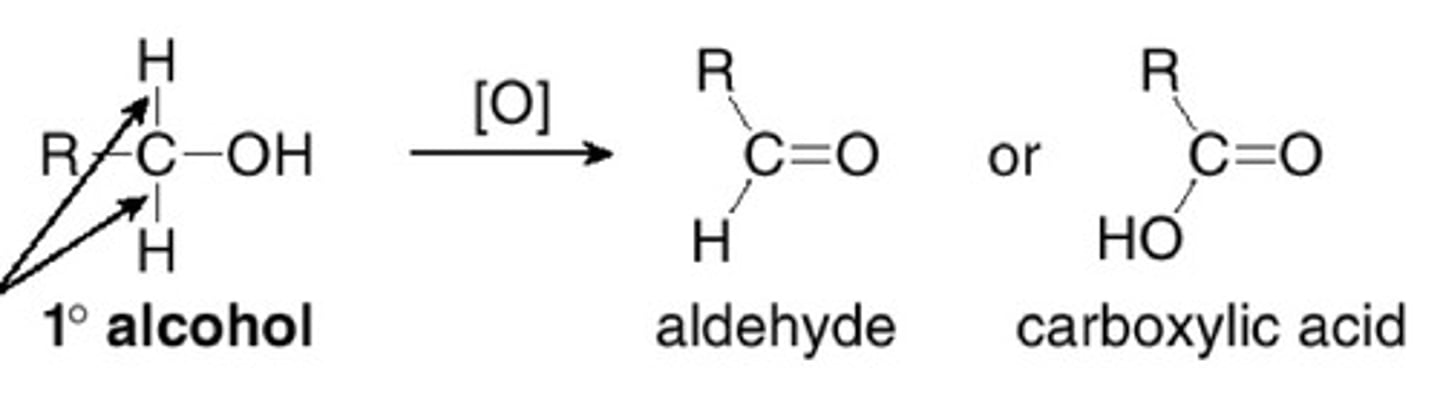
Oxidation of 1st Degree ROH to aldehyde
use PCC, CrO3, Py, HCl
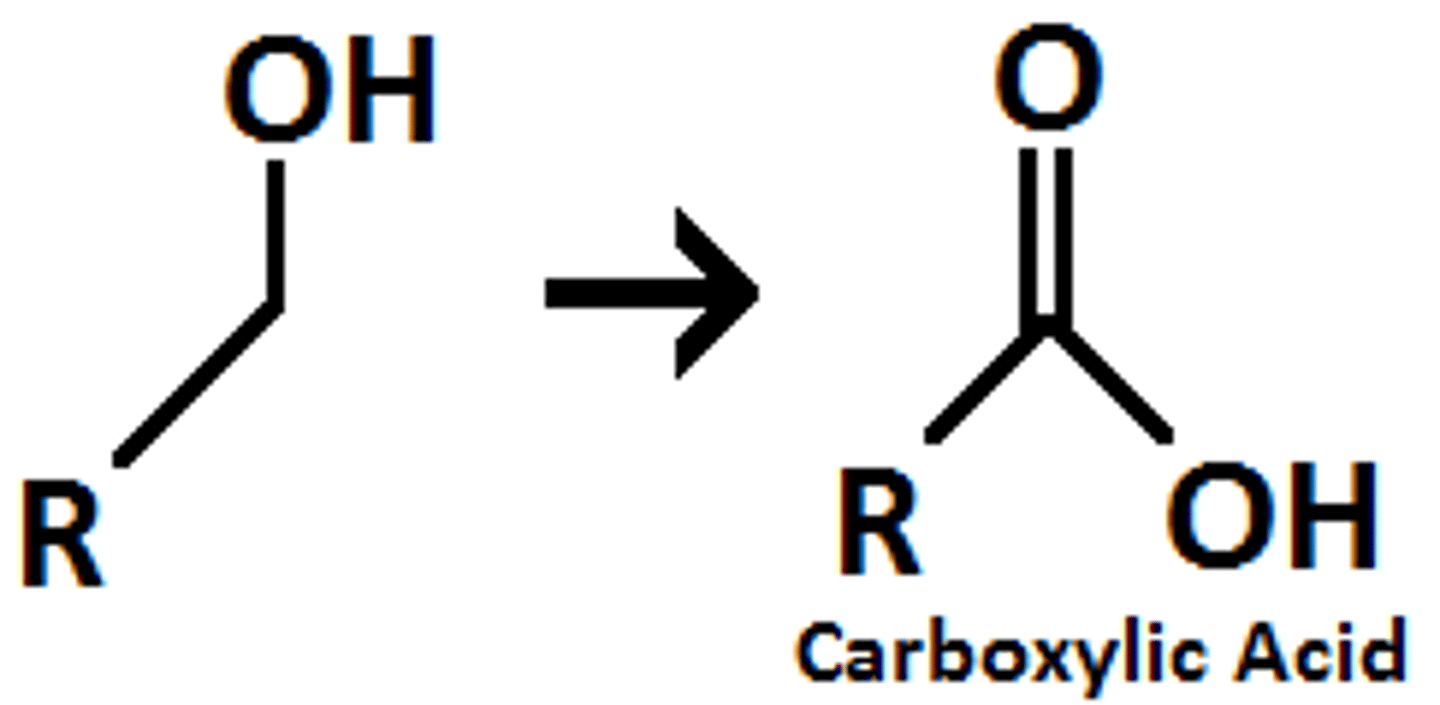
Oxidation of 1st Degree ROH to Carboxylic Acid
Strong, fully oxidized
Na2CrO4+H2SO4+H2O, KMnO4+HO-+H2O
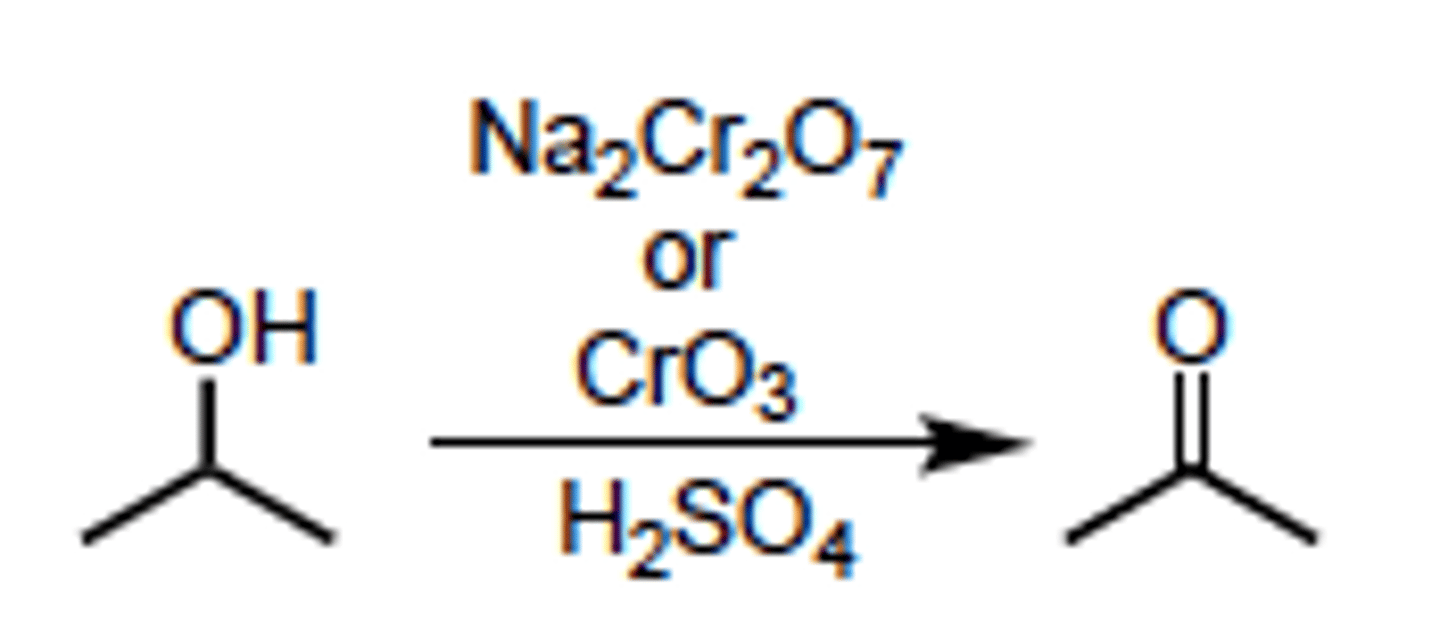
Oxidation of 2nd Degree Alcohol to Ketone
any Cr or Mn based reagent set
Synthesis of Linear Ethers
1) React alkoxide with alkyl halide (basic conditions via SN2)
2) Dehydrative Coupling of two alcohols (acidic conditions, SN2 or SN1)
3) Acid catalyzed addition of ROH to alkene (C+ intermediate with Markovinikov addition, racemic mix)
4) Alkoxymercuration-reduction addition of ROH to alkene (cyclic mercurium cation, Markovnikov addition, racemic mix-- RO attacks most sub'd C of cyclic cation)

ROR and H-X acid Reaction
ROR + 2 HX -> R-X + R'-X + H2O
heat required
1st degree C(alpha) = SN2
3rd degree C(alpha) = SN1 with C+ intermediate
Epoxide
Cyclic Ether, nucleophilic addition relieves ring strain (substitution)
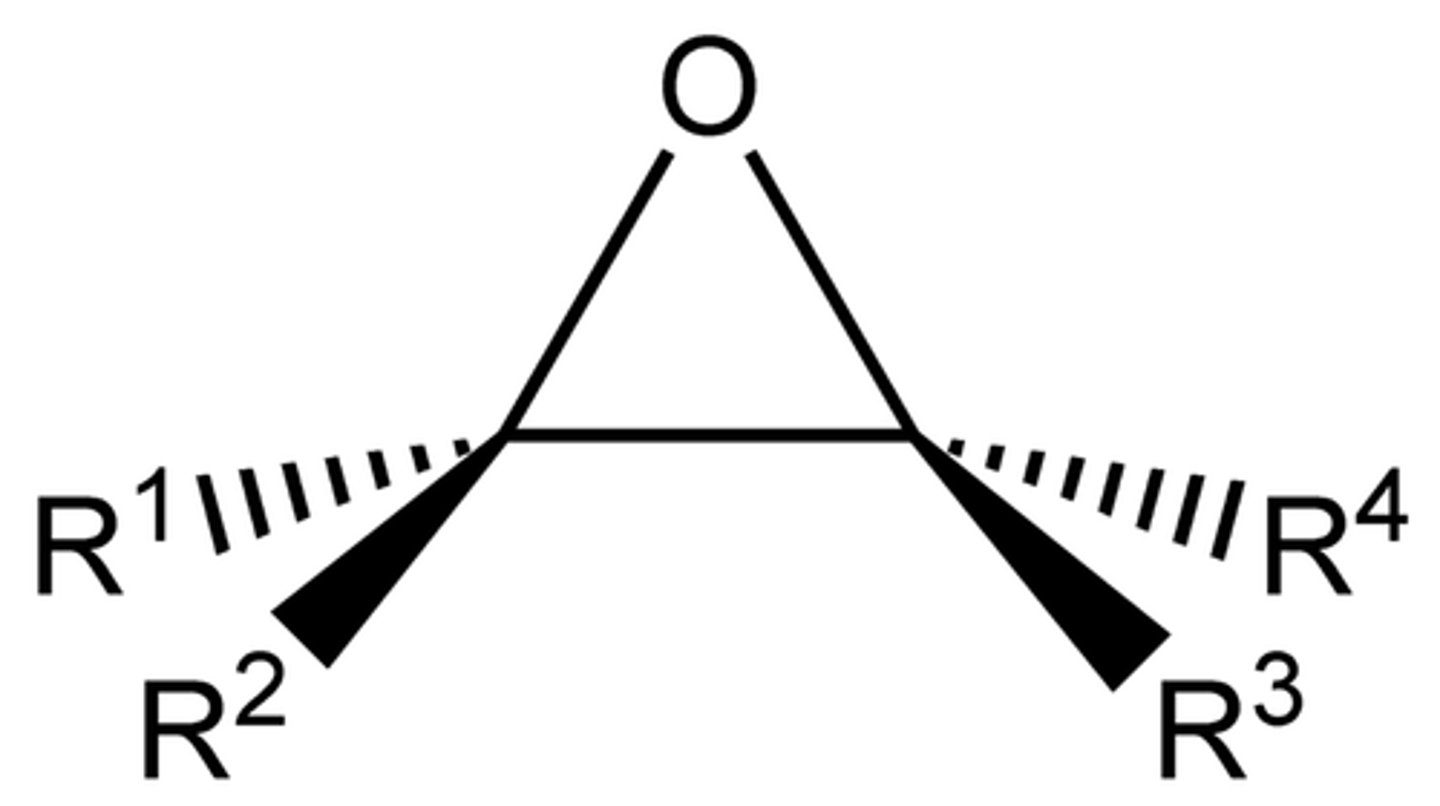
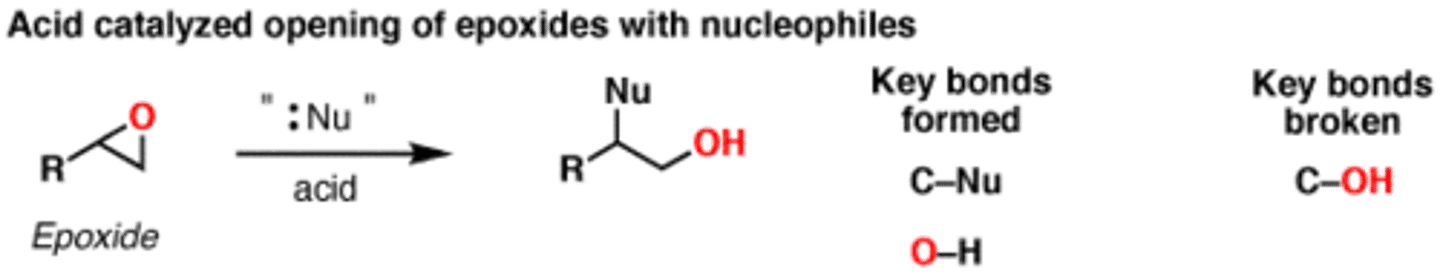
Opening Epoxides under Acidic Conditions (H-Nu:)
H2O: trans-1,2-diol formation
ROH: trans-1,2-hydroxy-ether formation
SN2 based mechanism
Regiospecific: incoming HO or RO adds to most-sub'd C of ring
Stereospecific: anti formation of new HO group
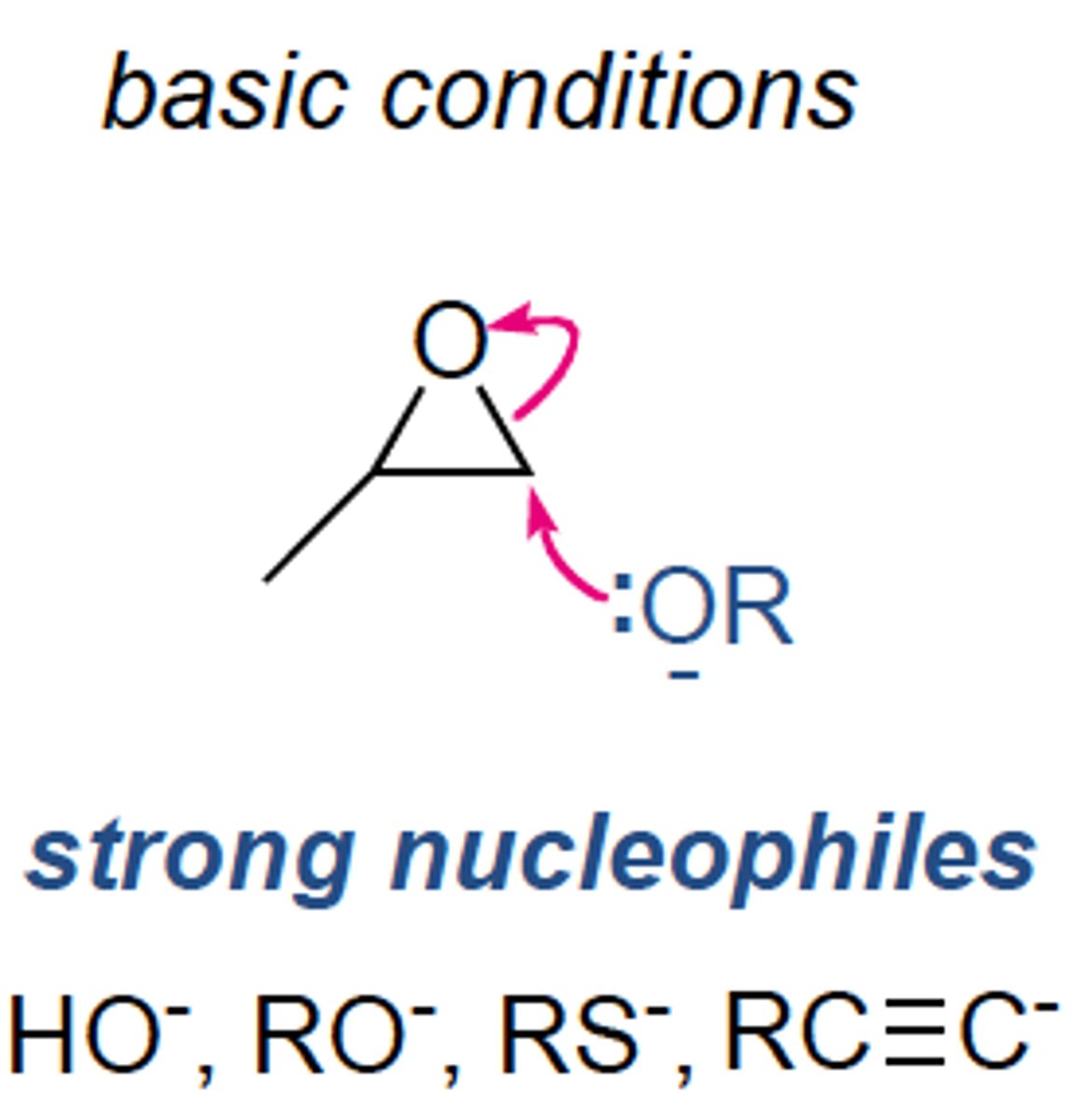
Opening Epoxides Under Basic Conditions (Nu:-)
OH- followed by neutralization: trans-1,2-diol formation
Other Nu:-: same as above
Regiospecific: Nu:- adds to least sub'd C of ring
Stereospecific: anti formation of HO group and Nu:-
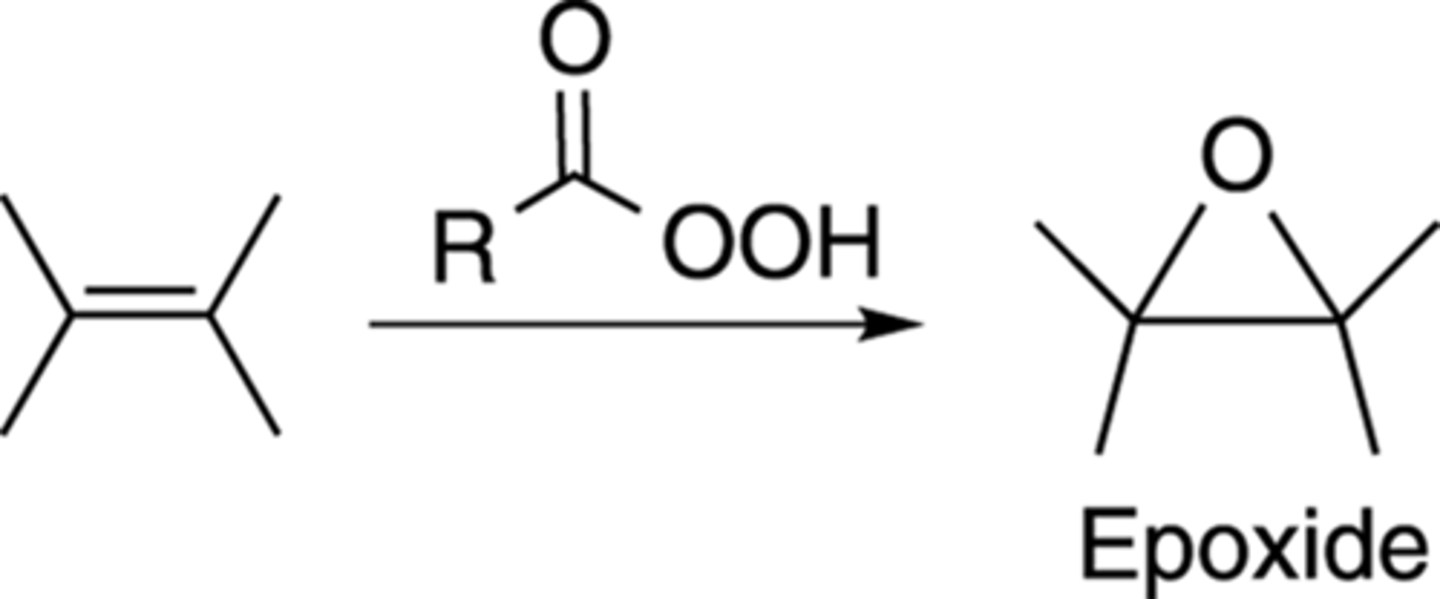
Synthesis of Epoxides from Alkene + Peroxy Acid
(RC(O)OOH) under acidic conditions forms epoxide ring
Regioselective: n/a, bridges both C
Stereospecific: syn addition of O to flat C=C bond (but can add top or bottom)
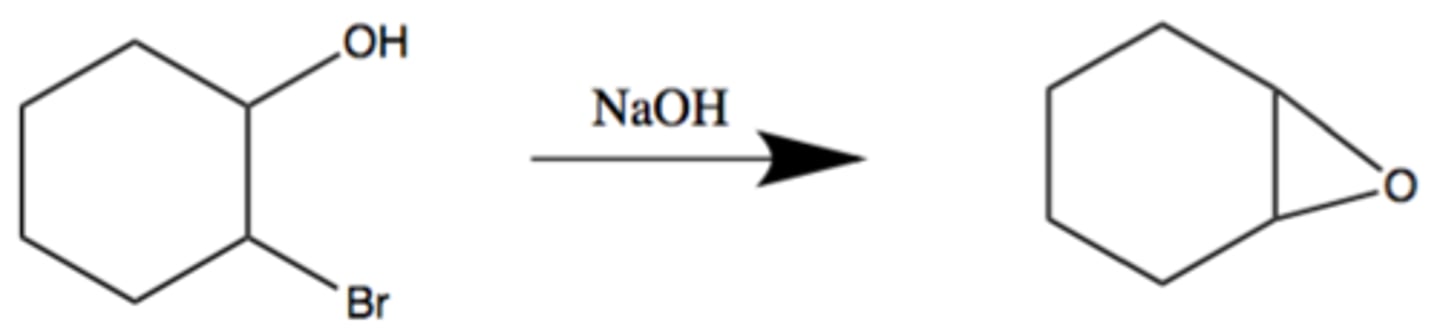
Synthesis of Epoxide from 1,2-halohydrin
Basic conditions
SN2 based INTRAmolecular cyclization rxn: ROH deprotenates to form ring
Regiospecific: formed O- adds to C(alpha) of C-X bond
Stereospecific: inversion of C(alpha) group