Homogenous Catalysis
1/28
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
29 Terms
Outline the pros and cons of homogenous and heterogenous catalysis
Homogenous:
Advantages: high rates, robust to poisons, highly selective, mild conditions, catalyst tuning, easier to probe mechanism.
Disadvantages: difficulties in separation, recovery and expensive.
Heterogenous:
Advantages: readily separated and recycled, long-lived, cheaper
Disadvantages: lower rates, sensitive to poisons, lower selectivity, poor mechanistic understanding, higher energy conditions.
Outline the Sabatier principle
A good catalyst must bind reaction intermediates neither too strongly or too weakly.
Too weak: reactants do not adsorb effectively, so little reaction occurs.
Too strong: intermediates get stuck on the surface and block active sites.
→ volcano plot of catalysts.
Outline the Sheldon E factor → what is this used to show?
E factor = mass (kg) of waste / mass (kg) of product. Measure of how environmentally efficient a chemical process is i.e. how much waste is produced.
Give equations for TON and TOF - what is each measuring?
TON = number of reactant molecules converted (or moles of product produced) per catalytic site. Measures productivity of a catalyst.
TOF = number of reactant molecules converted (or moles of product) per catalytic site per unit time. Measures activity of a catalyst.
Give the equation for selectivity of a catalyst, and define the lifetime.
Selectivity (%) = (moles of reactant converted to product)/(moles of reactant converted to all products) x100.
Lifetime: time for which catalyst maintains a 'sufficient' level of activity / selectivity.
Why is a homogenous catalyst required for hydrogenations of olefin/carbonyl?
H2 cannot undergo concerted reaction with alkene due to the symmetry of the H2 bond, hence will not have the correct symmetry overlap at both hydrogens.
Homogenous catalyst can break the H-H bond symmetry by forming a cis di-hydride, allowing interaction.
Can also employ chemoselectivity (terminal alkenes preferred) and regioselectivity and stereoselectivity (asymmetric reduction)

Explain why Wilkinson’s catalyst is a good catalyst in homogenous hydrogenation
Operates at mild conditions.
Uses an electron rich Rh(I) centre.
PPh3 is strongly sigma-donating, increasing electron density at Rh(I) to facilitate oxidative addition of H2.
PPh3 can be readily lost due to steric competition → 14 electron Rh(I) species that is electron deficient, allowing solvent to attach. Coordinatively unsaturated, allowing oxidative addition of H2.
How is wilkinson’s catalyst formed? What conditions does this formation take place under?
RhCl3 + 4PPh3 + H2O → Rh(Cl)(PPh3)3 + Ph3P=O + HCl + 2H2O
Uses solvent (EtOH) to prevent dimerisation → inactive complex. Must be solvent that acts as a hard ligand t.f. easily displaced by alkene.
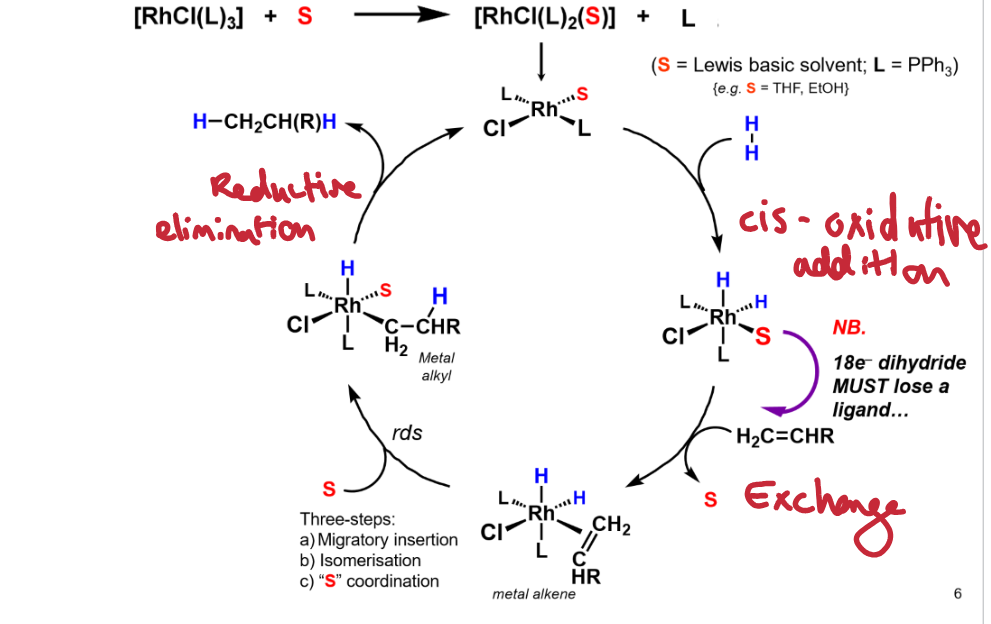
Give the catalytic cycle for Rh catalysed hydrogenation
Step 1: cis oxidative addition.
Step 2: ligand exchange
Step 3: migratory insertion, isomerisation, S coordination.
Step 4: reductive elimination.
Done in the presence of a solvent to prevent dimerisation of catalyst
Outline the chemoselectivityand regioselectivity in homogenous olefin hydrogenation
Will attack least substituted, terminal alkenes first.
Alkynes before alkenes, and alkenes before carbonyls/nitriles/NO2/aromatics, which are not reduced usually under typical Rh hydrogenation conditions
H2 addition to alkene is syn i.e. adds to the same face of the alkene.
How can the phosphine ligand be used to tune reactivity of the catalyst?
Most donating phosphines (lower CO stretching frequency in Ni(CO)3(PR3)) promotes alkene/hydride migratory insertion and oxidative addition of H2 leading to increased reactivity by making the Rh centre more electron rich.
Note: if too donating e.g. when R = alkyl, they won’t be displaced by the alkene so won’t react
How can we use chiral phosphine ligands to control H2 attack → enantiomeric excess in products.
Diphosphine = strongly chelating so remain coordinated throughout the whole reaction mechanism so that Rh remains in a chiral environment.
Example: BINAP, DIOP, CHIRAPHOS, DUPHOS.
Leads to high enantiomeric excess in products because during migratory insertion the complex is forces to transfer hydrogen to only one of the two possible enantiotropic faces of the alkene.
Uses bulky phosphine groups where if one side of the alkene is more sterically demanding, it will favour coordination away from bulky phosphine groups. (QUADRANT MODEL)
Outline donor-assisted Rh-catalysed hydrogenation
Alkene contains a donor group e.g. HC=O, OR, NR2, O-CNR2 which the lone pair of heteroatom can bind to the Rh centre, coordinating alkene at a fixed orientations → controls stereochemistry of hydrogenation by controlling face of alkene approach.
Give the general equation for olefin hydroformylation + its two products.
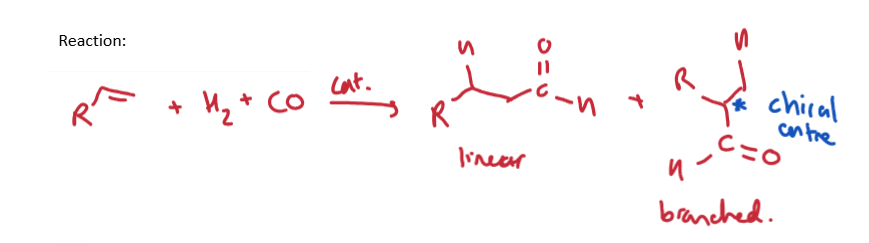
Give the general catalyst composition for olefin hydroformylation
HxMy(CO)z(L)n - i.e. must feature a metal carbonyl hydride species.
What were the problems with the first generation process using HxCoy(CO)2
High pressure and temperature
Not very good l:b ratio (3:1)
Hard to separate product from catalyst - has to be removed using NaHCO3 prior to distillation.
Give the catalytic cycle for the first generation process.
Oxidative addition of alkene cis to H due to trans influence of H + sterics.
Migratory insertion of H, adding to carbon closest to it →branched or linear. (Linear = favoured, less steric clash in coordination sphere)
New CO binds, inserting into the C-Co bond (migratory insertion)→ empty coordination site.
Reaction with hydrogen (OA + RE) to produce aldehyde and regenerate the catalyst.
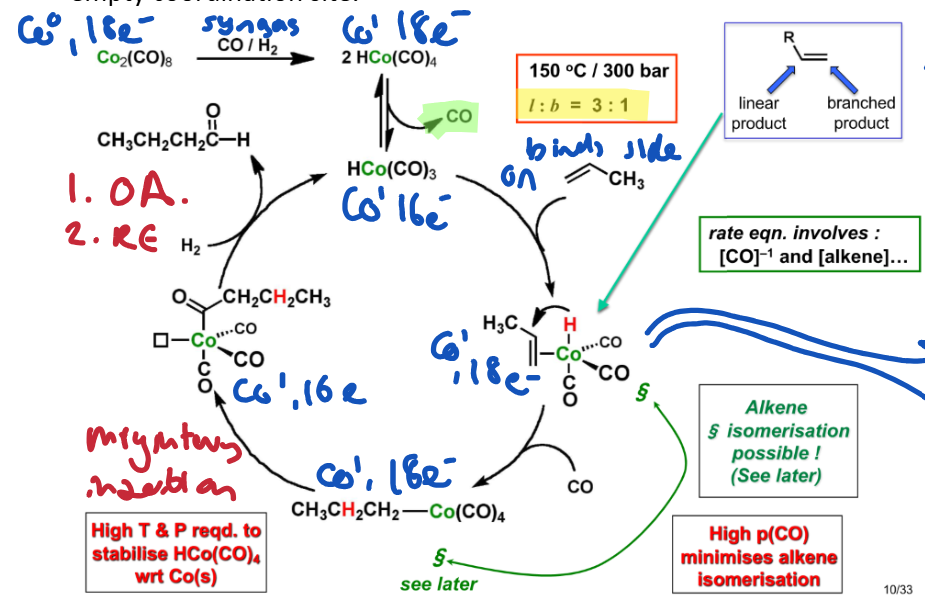
Explain why rate is proportional to 1/[CO] for olefine hydroformylation, and why we use high p[CO]
Leads to lower rates of reaction as too much CO can block coordination sites on Rh from alkene binding.
However, high p[CO] drives migratory insertion of hydride, and prevents H2 elimination (which can then lead to decomposition to inactive metal form) hence stabilises the active Rh catalyst.
How did switching to phosphine ligands improve the catalyst in olefin hydroformylation?
Phosphine = better Lewis base, stronger binding to the metal.
Stabilises the catalyst by preventing degradation to Co metal.
Lowers pressure and temperatures.
Improves l:b (9:1) - makes H more hydridic in character, favouring anti-Markonikov addition → linear product.
Stronger sigma donor → improved Co→CO backbonding, making CO dissociation harder.
(H, slowed rates and activated towards olefin hydrogenation → alcohol product).
How did swtiching to Rh improve the catalyst for olefin hydroformylations?
Lowered T and P even further.
Further increase in l:b (92:8).
Greater activity by favouring C-Rh bond formation.
Stable catalyst, can undergo product distillation.
Give an equation for preparing the Rh pro-catalyst, HRh(CO)(PR3)3
E.g. Rh(acac)(CO)2 + H2 + CO + 3PR3 → HRh(CO)L3 + Hacac + 2CO
Give the catalytic cycle for Rh/PR3 catalysed olefin hydroformylation
Alkene added and CO removed (ligand exchange).
Migratory insertion of H
Migratory insertion of CO.
Oxidative addition of H2
Reductive elimination.
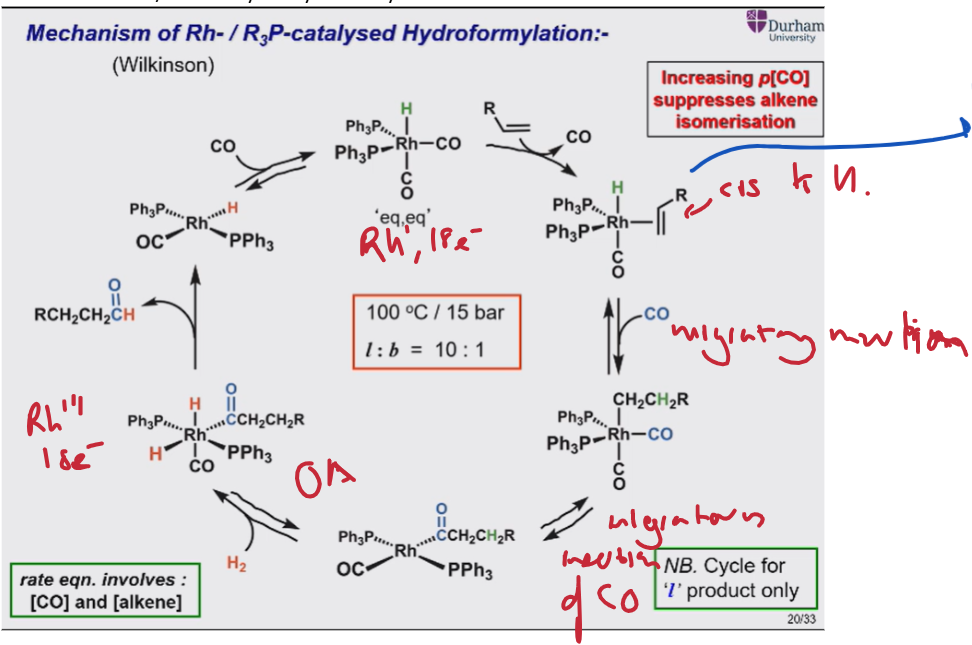
Outline the ways in which the phosphine can be modified to favour the linear product.
More basic phosphine = more electron donating → improve l:b ratios as favours H- (hydridic) character. H, disfavours CO dissociation which can lower rates.
Bidentate phosphine with bite angle close to 120 degrees. Favours eq,eq phosphine → linear product by forcing CO trans to H → more hydridic H- character. As well as sterically favouring the correct orientation of alkene → linear product.
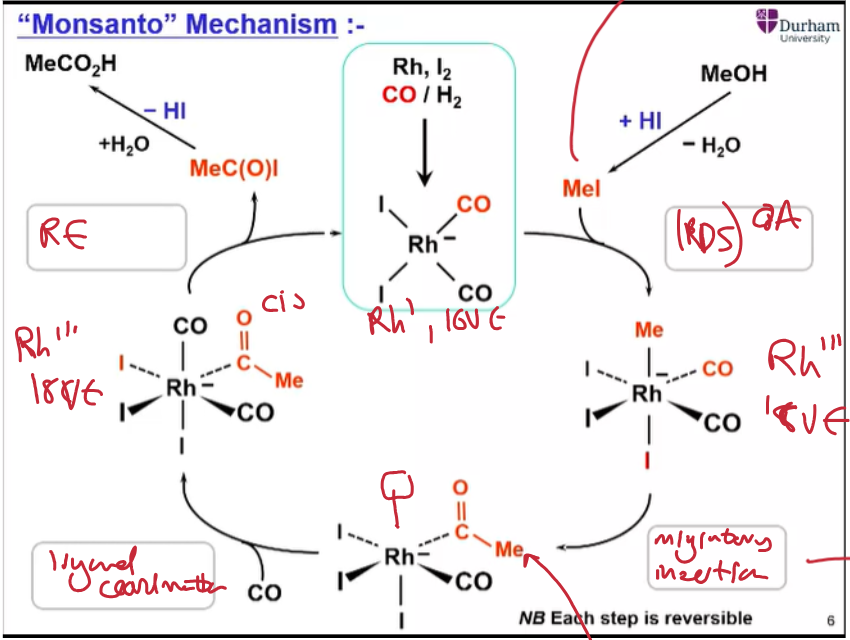
Give the catalytic cycle for acetic acid manufacturing using the Rh,I2 catalyst.
Oxidative addition of MeI (limited reagent by controlling HI addition). RDS.
Migratory insertion of methyl onto CO.
Ligand coordination of CO.
Reductive elimination, followed by hydrolysis to get product.
Why is the concentration of MeI controlled?
Very volatile and toxic reagent
Why is iodine used over other halogens?
Rate of reaction depends on oxidative addition, which is essentially a SN2 process of Rh- attacking and kicking out I-.
I- = good LG (weak C-I bond) so fast process.
I- = soft ligand, favouring Rh(I) (soft metal), stabilising catalyst.
Why do we use inorganic iodide promotors in acetic acid manufacturing?
Stabilises the Rh species
Lowers [HI] by providing an alternative, less reactive source of I- (since HI can react with the catalyst to form an inactive species).
Allows lower water concentrations to be used (that usually lower [HI] through conversion, but also → water-gas-shift reactions and requires more product drying.
What is one problem with using iodide promoters?
iodide salts are very corrosive – expensive alloy materials used to prevent corrosion of reaction materials
Why might one switch from Rh → Ir in acetic acid manufacturing?
Cheaper.
Limits WGS since less water required to stabilise - less CO → CO2
-> purer acetic acid by limiting WGS and therefore formation of H2 → side reactions.
Second row -> third row = stronger M-C bond strength for Ir so can use much less [MeI] in the OA RDS step (faster oxidative addition step)
H, strong Ir-C bond also hinders RE step. Need to add iodide promoter to achieve a good rate of reaction.