MED CHEM EXAM 3
1/302
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
303 Terms
Role of Aspirin
Pain ↓
Fever ↓
Inflammation ↓
Prevents clots (low dose)
MOA of Aspirin
Irreversibly inhibits COX enzymes
→ ↓ prostaglandins
What causes symptoms (pain, redness, swelling)?
Prostaglandins (pain + fever)
Cytokines (like TNF, IL-1, IL-6)
Histamine (vasodilation)
These cause:
↑ blood flow → redness, heat
↑ leakage → swelling
↑ nerve sensitivity → pain
Acute vs Chronic Inflammation
Acute (short-term)
Fast, resolves
Cells: neutrophils
Example: cut, infection
Chronic (long-term)
Slow, damaging
Cells: macrophages, T cells
Leads to diseases like:
Arthritis
Diabetes
Heart disease
Cancer
Aspirin MOA
MOA: Irreversibly inhibits COX enzymes → ↓ prostaglandins
Uses: Relief of minor pains, fever, rheumatologic disease symptoms, and prevention of a MI.
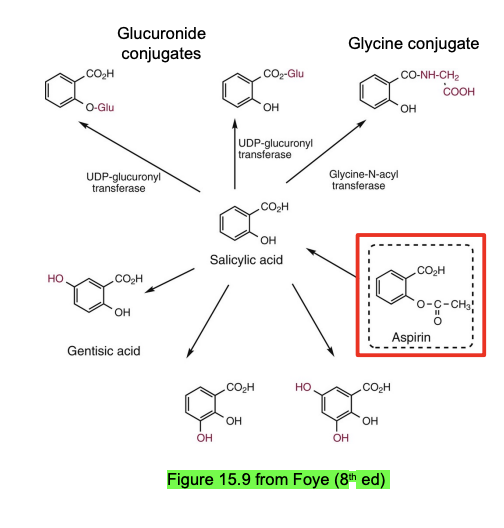
Rapidly de-acetylated to salicylic acid → further glucuronidated + glycine to be excreted.
CI for active GI bleeds, hepatic impairment, post flu infection.
Aspirin’s therapeutic effects follow its inhibition of the ____ known cyclooxygenase enzymes
3
COX-1
Constitutive (“always on”) enzyme involved in normal body functions (housekeeping)
Ubiquitous within tissues (Especially platelets, stomach, kidneys).
Stimulates the synthesis of PGs needed for tissue maintenance
Downstream: Protects stomach lining, maintains kidney blood flow, promotes platelet aggregation
COX-2
Inducible enzyme activated during inflammation
Induced at sites of inflammation (blood vessels, fibroblasts, endothelial cells)
Causes inflammation, pain, and fever
Aspirin effect on COX-2: Weaker than COX-1 (~100x less potent).
COX-3
Variant of COX-1 mainly in the brain
Function: Likely involved in pain and fever (not fully understood)
Aspirin effect on COX-3: Weak
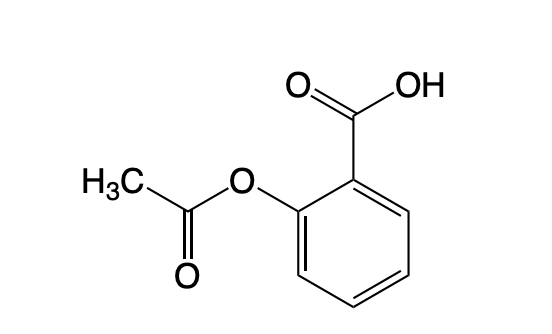
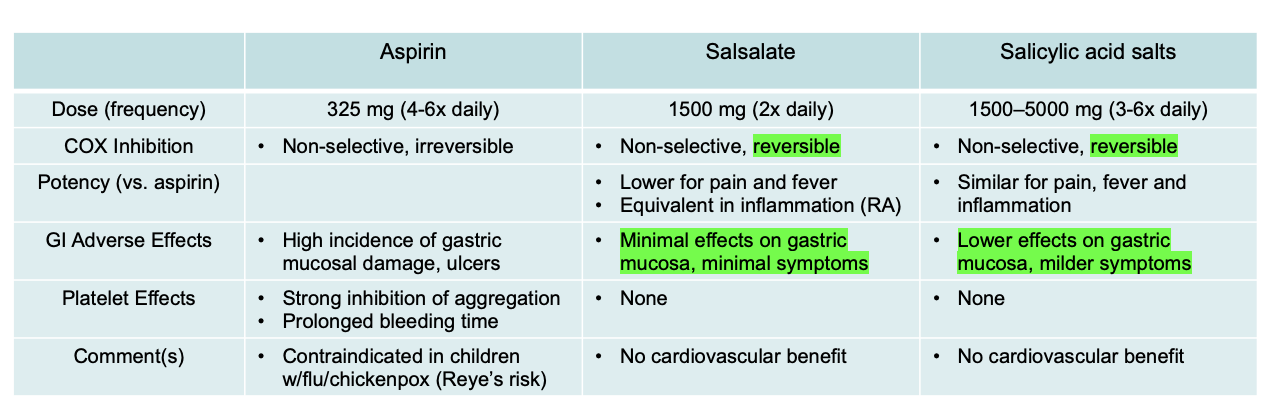
Aspirin
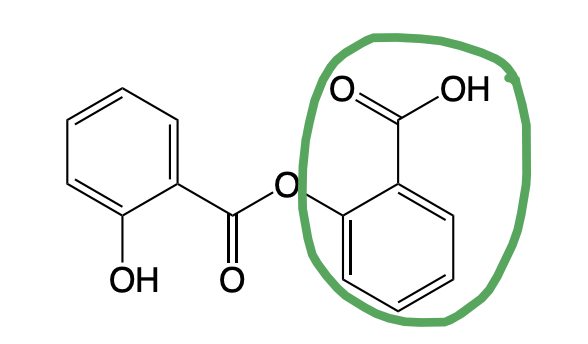
Salsalate
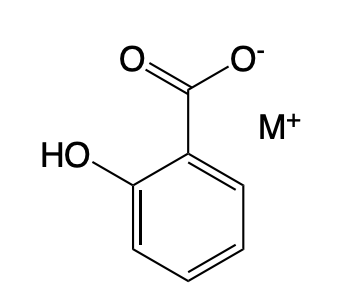
Salicylic Acid Salts
Aspirin ADME
Absorption
Oral BA → Affected by food, pH, formulation, gastric emptying, antacids
Metabolism
Rapidly converted → salicylic acid (plasma esterases)
Salicylic acid → glycine (75%) + glucuronide (15%) conjugates by Phase II metabolism
Excretion
Kidney (urine): salicylic acid + metabolites
CI of Aspirin
Active GI bleeding
Severe liver or kidney disease
Children post-viral infection (flu/chickenpox) → risk of Reye syndrome
Adverse effects / precautions of Aspirin
GI irritation, ulcers, bleeding (↑ risk in elderly, alcohol use, GI history)
Increased bleeding risk (surgery, dental work, anticoagulants)
May trigger asthma or allergic reactions (incl. anaphylaxis)
Reye syndrome (rare but serious brain + liver damage)
Pregnancy: potential fetal/infant risk
DDIs of Aspirin
Other NSAIDs, antiplatelets, corticosteroids → ↑ GI/bleeding risk
Anticoagulants (e.g., warfarin) → ↑ bleeding risk (↑ free drug)
Methotrexate → ↓ renal clearance → ↑ toxicity
Diuretics/aldosterone antagonists → ↓ effectiveness (↓ prostaglandins)
Aspirin History
Ancient: Willow bark used for pain/fever (>3500 years)
1800s: Salicylates identified → aspirin developed; GI toxicity noted
MOA discovered (1960–80): COX inhibition → ↓ prostaglandins & thromboxane
Modern use: Pain/fever + antiplatelet (↓ MI, stroke, TIA); possible cancer & preeclampsia benefits
Salsalate
Salicylic Acid Analog
Reversible Inhibition of COX
Less potent for pain and fever but equivalent for inflammation (RA) as Aspirin
Minimal effect on the gastric mucosa → less damage than Aspirin
No CV Benefit
Salicylic Acid Salts
Salicylic Acid Analog
Reversible Inhibition of COX
Similar effect for pain, fever, and inflammation as Aspirin
Lower effect on the gastric mucosa (than Salsalate) → even less damage than Aspirin
No CV Benefit
Aspirin vs Salicylic Acid Analogs
Diflunisal
Mechanism: Reversible non-selective COX inhibitor, more potent than Aspirin, less platelet inhibition
Use: Symptomatic relief of mild–moderate pain, osteoarthritis, and rheumatoid arthritis (same).
ADR: Same GI risk but better GI tolerance, no risk for Reyes, CI for HF patients.
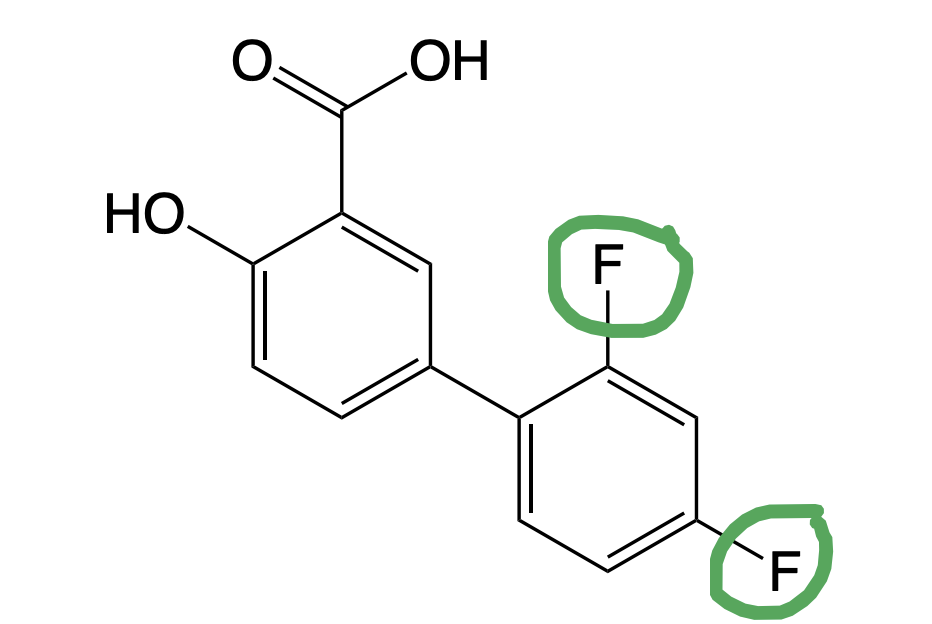
Diflunisal
→ Di fluoro
Acetaminophen Uses
Pain: headache, cold, toothache, backache, menstrual cramps
Fever reduction
Similar efficacy to aspirin for pain/fever
No anti-inflammatory or anti-platelet effects
MOA of Acetaminophen
Weak central COX peroxidase inhibition → ↓ CNS prostaglandins
↑ serotonergic descending pain pathways
↑ endocannabinoid + TRPV1 activity → analgesia
CIs for Acetaminophen
Allergy to drug
Severe liver disease
Warnings of Acetaminophen
Overdose → severe hepatotoxicity → liver failure/death
Caution: liver disease, alcohol use, malnutrition, renal impairment
Stop if rash or hypersensitivity occurs
DDIs of Acetamiophen
CYP2E1 inhibitors → ↑ toxicity risk (hepatotoxicity)
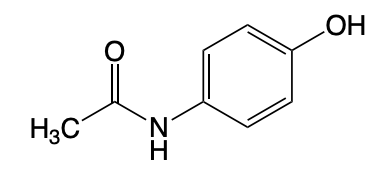
Acetaminophen
Acetaminophen
Use: Relief of minor aches and fever. (Difference from Aspirin → no antiplatelet/antiinflammatory effect)
MOA: Inhibits COX enzymes, activates serotonergic inhibitory pathways, and activations endocannabinoids in the CNS.
CI: Hypersensitivity to Acetaminophen + Hepatic Impairment
CAN BE SUBSTITUTED FOR ASPIRIN FOR PAIN RELIEF/FEVER REDUCTION IN PATIENTS WITH GI ISSUES AND RISK FOR REYE’S SYNDROME
DDI: CYP2E1 inhbiitors
Advantage of Acetaminophen > Aspirin
Safe alternative to aspirin in:
GI bleeding risk
Bleeding risk patients
Children with viral illness (no Reye syndrome)
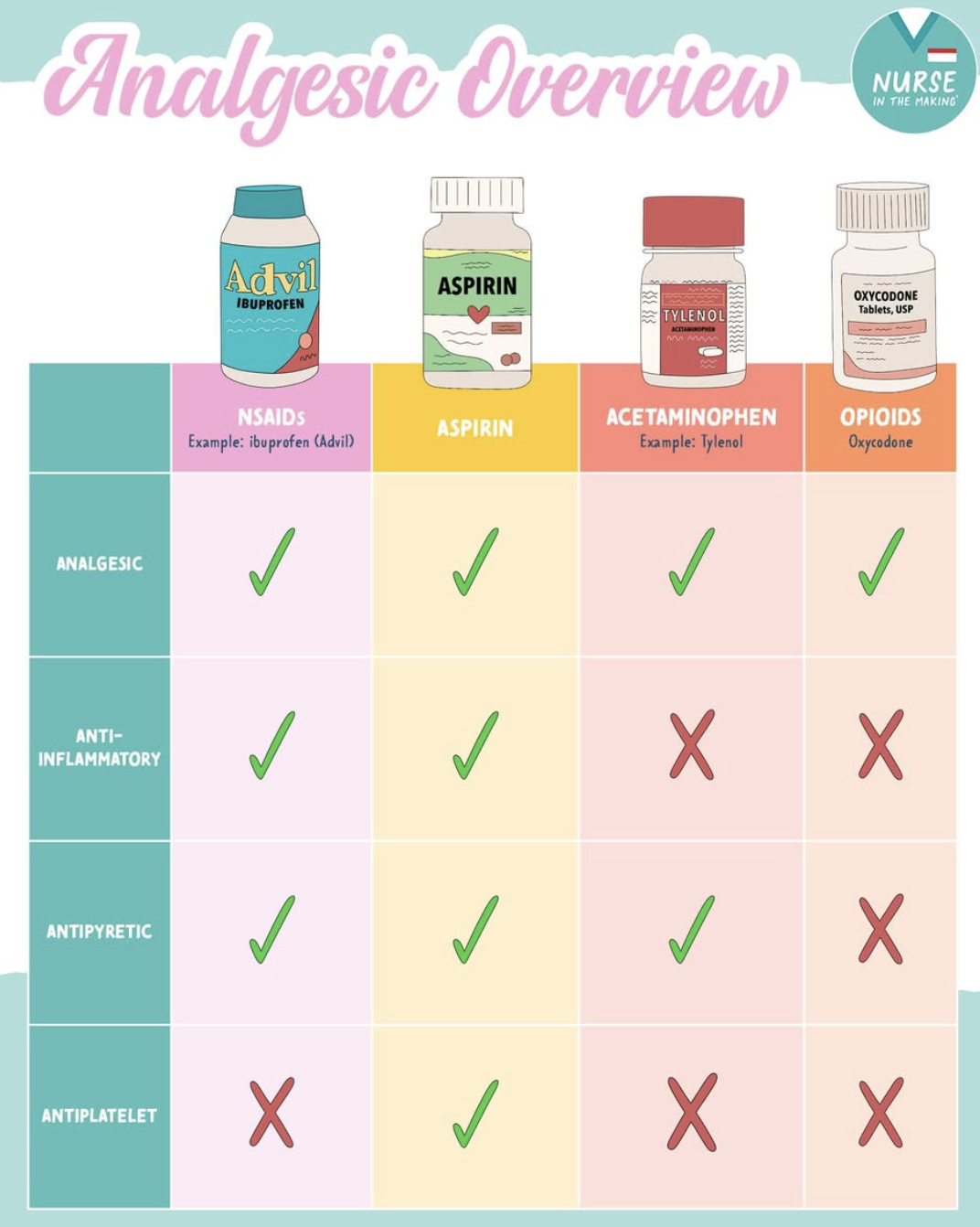
ADME of Acetaminophen
Metabolism (main)
Phase II conjugation: Glucuronide & Sulfate
Minor toxic pathway
CYP2E1 + CYP3A4 → NAPQI (toxic metabolite)
Normally detoxified by glutathione
Overdose → glutathione depleted → liver toxicity
Alcohol interaction
Ethanol ↑ CYP2E1 → ↑ NAPQI → ↑ hepatotoxicity risk
Brain metabolism
Deacetylation + conjugation with arachidonic acid (CNS-related pathway)
Pharmacologic Consequences of COX Inhibition
COX-1/2: Homologous Enzymes that convert Arachidonic Acid → Prostaglandin H2
Differ in regulation, tissue distribution, and predominant physiologica/pathophysiologic roles
Clinical implication
COX inhibition → ↓ prostaglandins → ↓ pain, fever, inflammation, and/or protective functions depending on COX type inhibited
Aspirin’s Effect on Eicosanoids
PGs → Reduces inflammation and pain by decreasing PG synthesis
Thromboxanes → Reduces the risk of stroke/heart attack by decrease TX synthesis
Decrease in PG and TX synthesis → Increases LT synthesis
Leukotriene → 3 Conjugated DB
What are the functions of Prostaglandins?
Smooth Muscle → Can dilate or constrict vascular or uterine smooth muscle
Inflammation → Pro-inflammatory; Pro-nociceptive
Promotes Pain and Fever Response
→ PGE1 is important for GI protection and it’s synthesis is mediated by COX-1 (Main COX Enzyme blocked by Aspirin, less PGE1 → less cytoprotection of the GI → more GI effects as seen w/ Aspirin).
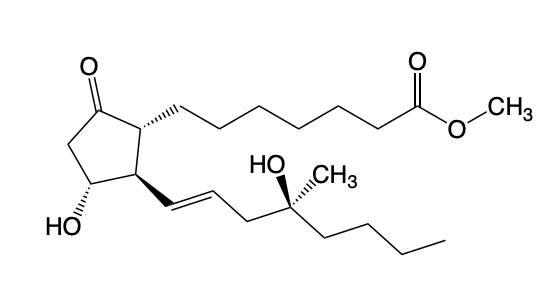
Misoprostol
→ Class: Prostaglandin Receptor Modulator Therapeutics
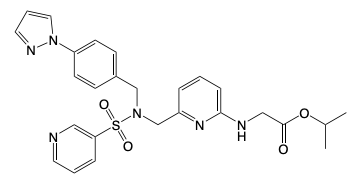
Omidenepag
→ Class: Prostaglandin Receptor Modulator Therapeutics
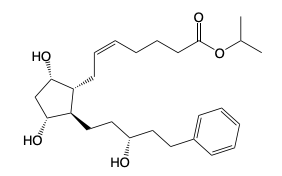
Latanoprost
→ Class: Prostaglandin Receptor Modulator Therapeutics
Eicosanoids: ____
Prostaglandins, Thromboxanes and Leukotrienes
→ Derived from AA
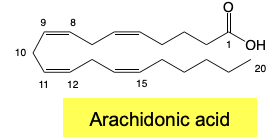
PG Structure SAR
5-membered ring (PENTAGON); C13-14 DB, C-15 Alpha OH
Subclasses → Depend on Nature & Stereochemistry of C9,11 oxygens
Roles in inflammation, pain, fever, blood flow, and smooth muscle contraction.
Aspirin reduces inflammation and pain by decreasing PG biosynthesis.
Thromboxane SAR/Function
Contains a 6-membered ring; C13-14 DB, C-15 Alpha OH
Promotes platelet aggregation → Aspirin reduces the risk of stroke & heart attack by decreasing TX biosynthesis.
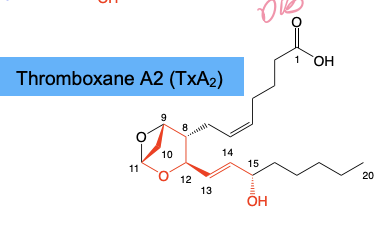
Prostacyclin & Thromboxane Biosynthesis
Selexipag
Selexipag
IP Agonist
Indication: Pulmonary HT
Leukotriene Functions
Pro-inflammatory
Lead to bronchoconstriction, leukocyte chemotaxis
Drugs that inhibit Leukotrienes: Montelukast, Zafirlukast, Zileuton
Montelukast
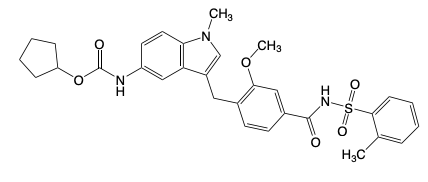
Zafirlukast
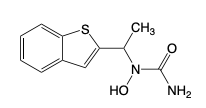
Zileuton
CC: Chemical Mediators
Prostaglandins: Pro-inflammatory, pro-nociceptive, promote pain & fever
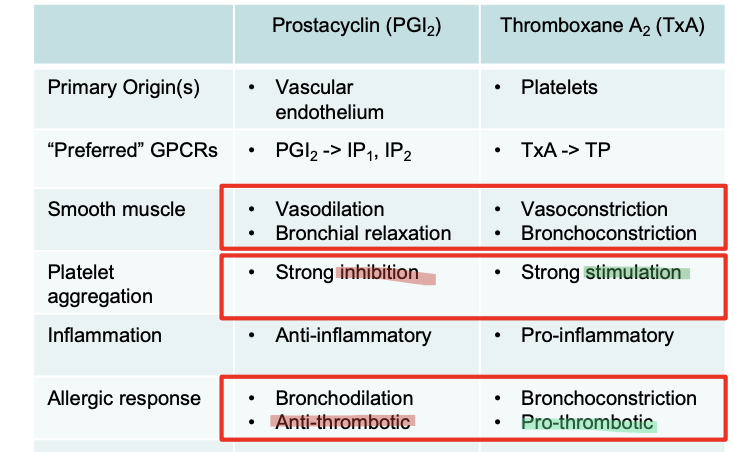
Prostacyclins (PG12): Vasodilation & bronchial relaxation
Thromboxane: Strong stimulation of platelet aggregation
Leukotrienes: Bronchoconstriction and Leukocyte Chemotaxis
Aspirin Effects on Chemical Mediators
Mechanism: Irreversibly inhibits COX → ↓ arachidonic acid products
↓ Prostaglandins (PGs): ↓ pain, ↓ fever, ↓ inflammation
↓ Prostacyclin (PGI₂): ↓ vasodilation + ↓ anti-platelet effect
↓ Thromboxane A₂ (TXA₂): ↓ platelet aggregation (key antiplatelet effect)
Leukotrienes: not blocked (can ↑ via shunting → bronchoconstriction risk)
Net effect: analgesic + anti-inflammatory + strong antiplatelet effect
Thromboxane vs Prostacyclin
Thromboxane: Keep them Together

NSAIDs (COX inhibitors) — COX-1 vs COX-2
COX-1 (protective enzyme)
Maintains stomach lining, kidneys, platelets
Inhibition → ↓ GI protection → ulcers/bleeding
Also → ↓ platelet aggregation (bleeding risk but cardioprotective in low-dose aspirin)
COX-2 (inflammatory enzyme)
Drives pain, fever, inflammation
Inhibition → ↓ pain/inflammation
BUT → ↓ prostacyclin (PGI₂) with intact TXA₂ → ↑ clot risk (MI, stroke), ↑ BP, fluid retention
SAR of COX Inhibitors
Core structure
Carboxylic acid (or bioisostere)
Short linker (n = 0–3, usually 1)
Aryl/heteroaryl ring(s)
Hydrophobic R groups
How they bind COX
Carboxylate binds Arg120
Aromatic rings do π–π interactions (Tyr, Trp)
R groups improve fit + potency
Key idea
Mimic arachidonic acid binding in COX active site
Selectivity
Most NSAIDs: non-selective COX-1/COX-2
COX-2 selective drugs (coxibs): designed to reduce GI toxicity while keeping anti-inflammatory effect
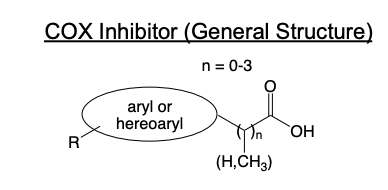
Classes of NSAIDs
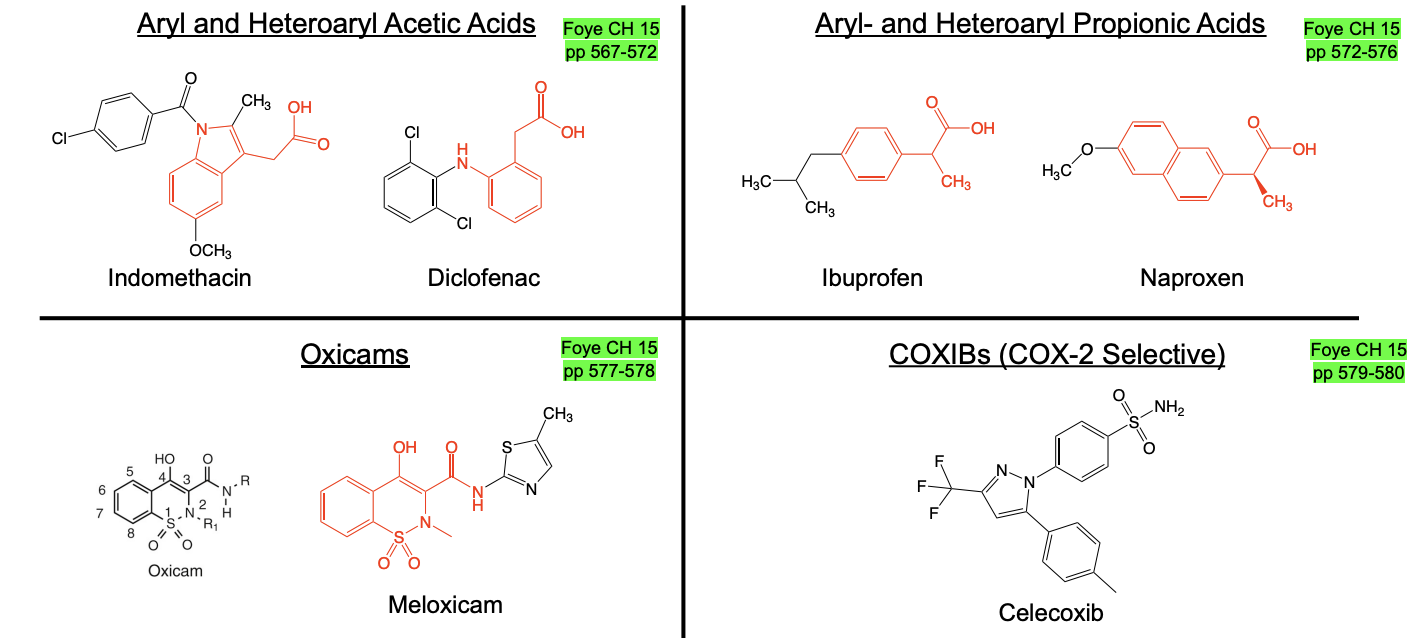
NSAIDs Impact on COX Enzymes
COX-1 blocked → ↓ stomach protection + ↓ platelets → ulcers + bleeding (± cardioprotection)
COX-2 blocked → ↓ pain/inflammation BUT ↓ PGI₂ → ↑ clot risk, ↑ BP, ↑ HF risk
Montelukast & Zafirlukast
Leukotriene Receptor Antagonist (Suffix: -lukast)
Indicated for asthma, exercise induced bronchoconstriction, allergic rhinitis
Zileuton
5-Lipoxygenase inhibitor
Indicated for asthma
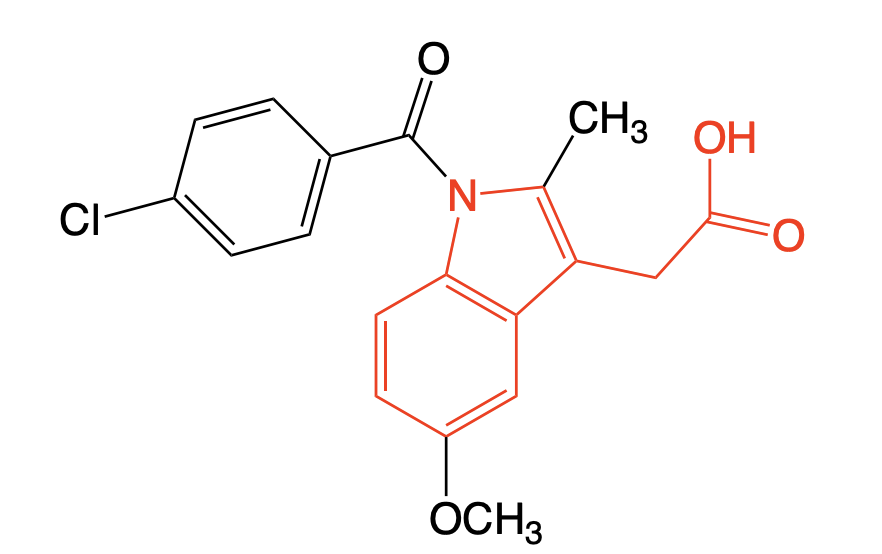
Indomethacin
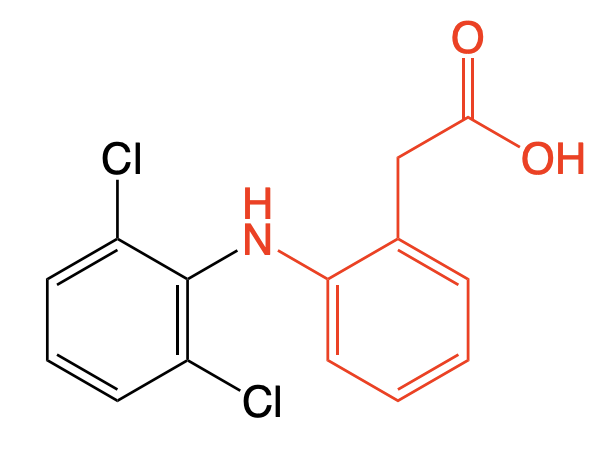
Diclofenac
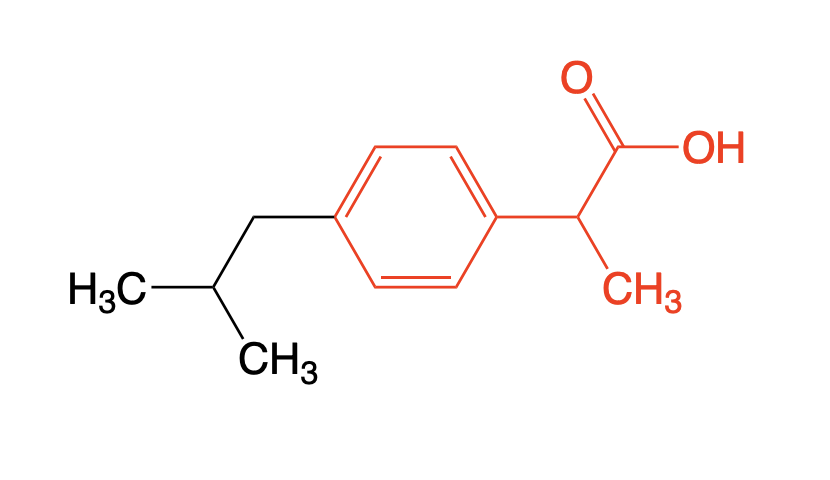
Ibuprofen
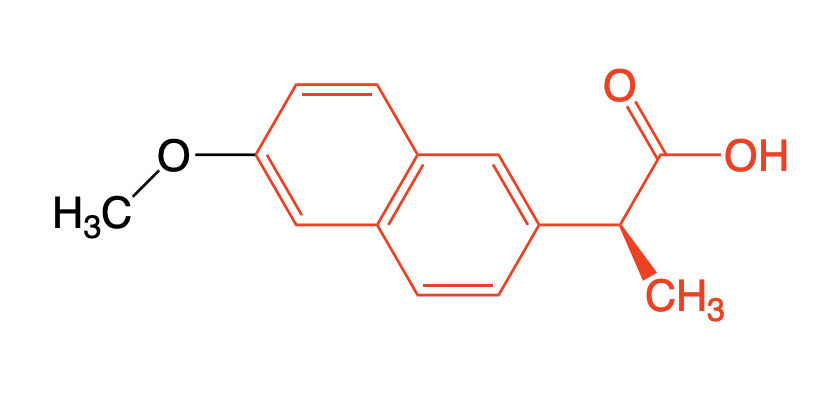
Naproxen
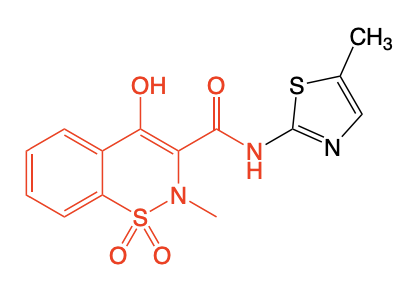
Meloxicam
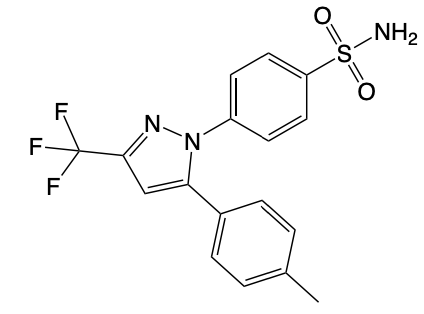
Celecoxib
Indomethacin
Class: Non-selective NSAID
MOA: COX inhibitor (pseudo-irreversible), more potent than Aspirin (analgesic and antipyretic activity).
Similar DDIs and effect of renal impairment as Aspirin
BBW: ↑ CV thrombotic risk (MI, stroke), especially post-CABG
Metabolism: Phase I Oxidation, Phase II Glucuronidation, Amide Hydrolysis → Inactive

Indomethacin SAR
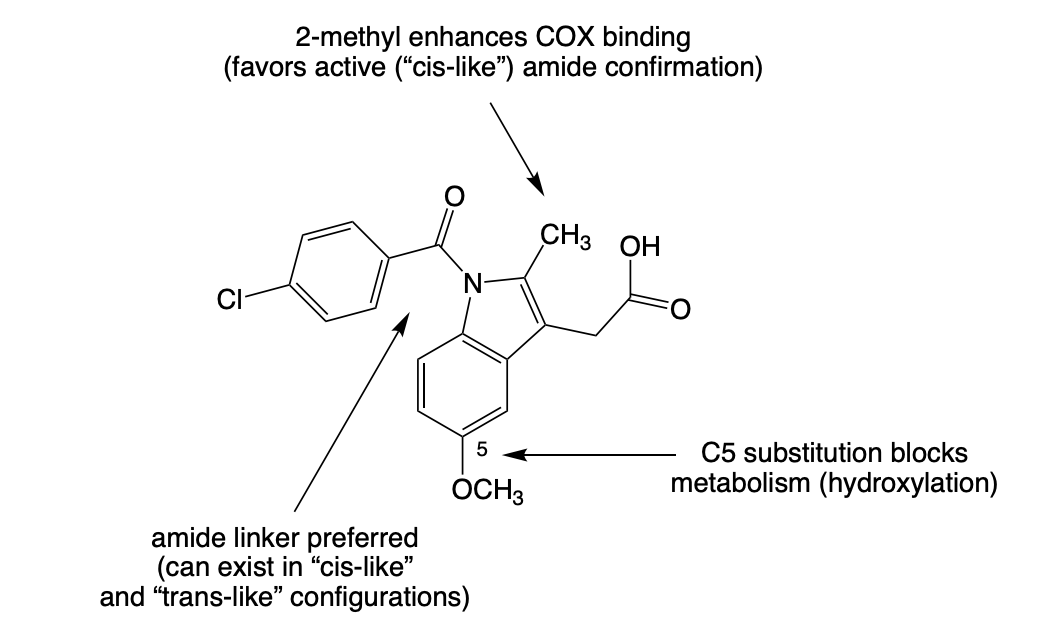
Diclofenac
Class: Non-selective NSAID with COX-2 preference
Also inhibits:
5-lipoxygenase
Phospholipase A₂
Very potent analgesic (~100× aspirin)
Use: Moderate-Severe Osteoarthritis, RA, and ankylosing spondylitis
BBW: ↑ CV thrombotic risk (MI, stroke); Contraindicated post-CABG
ADME: Phase I metabolism predominates, 4’ Hydroxy is the major metabolite → forms reactive quinoneimine.

Ibuprofen
Use: OTC for temporary relief from pain
MOA: Non-selective NSAID → equally potent w/ Aspirin
ADR: Similar to Aspirin but better GI tolerance
CI for use during pregnancy due to renal dysfunction and gestation disruption.
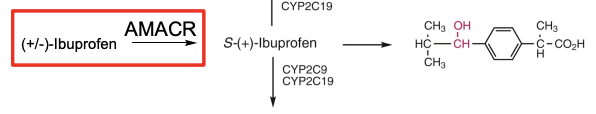
Ibuprofen ADME
Given as racemic mixture (R/S)
R → S conversion via AMACR → active form (S-ibuprofen)
S-form = pharmacologically active; R-form is largely inactive
Metabolism occurs before CYP oxidation to inactive metabolites
Naproxen
Use: Osteoarthritis, rheumatoid arthritis, ankylosing spondylitis
Mechanism: Non-Selective NSAID → Equally potent w/ Aspirin, Ibuprofen
Difference w/ Ibuprofen → Not a substrate for AMACR → GIVEN AS THE ACTIVE S ENANTIOMER.
Similar ADRs as Ibuprofen
Slightly ↓ CV risk vs ibuprofen
Slightly ↑ GI bleed risk (esp. with alcohol)
Meloxicam
MOA: “COX-2” preferring NSAID → 100x more potent than Aspirin, Ibuprofen
Use: Chronic Treatment (*DIFFERENCE*) of Osteoarthritis and Rheumatoid Arthritis
Once daily
BBW: CI for CABG (NSAID Class Effect); increased risk of CV events.
COX-1 vs COX-2 Inhibition — Summary
COX-1 (protective enzyme)
Maintains stomach lining, platelets, kidney blood flow
Inhibition → ↓ GI protection → ulcers, bleeding
↓ Platelets → ↓ clotting (beneficial in low-dose aspirin)
Can cause bronchospasm
COX-2 (inflammatory enzyme)
Drives pain, fever, inflammation
Inhibition → ↓ pain/inflammation
BUT ↓ prostacyclin → ↑ platelet activity + vasoconstriction
→ ↑ CV risk (MI, stroke), ↑ BP, ↑ heart failure risk
Celecoxib
Suffix: -coxib
SELECTIVE NSAID → for COX-2 (Inflammatory)
10x more potent than Aspirin/Ibuprofen for Arthritis Pain Relief
BBW: CI for CABG (NSAID Class Effect), analogous to Ibuprofen
More COX-2 selectivity = less GI harm but more CV risk
→ COX-2 Inhibitors are reserved for high risk GI patients.
NSAIDs: Difference
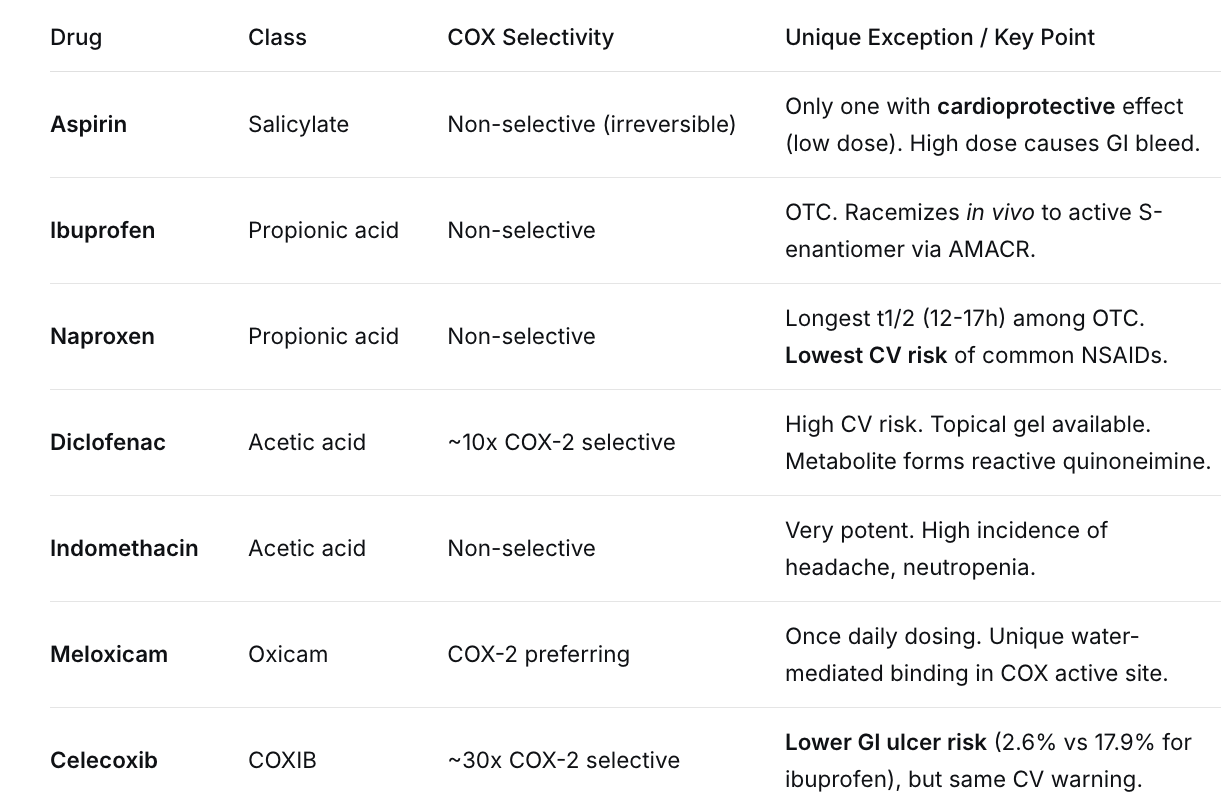
NSAIDs w/ lowest ADR RIsk
Naproxen → Low CV Risk
Celecoxib → Low GI Bleed Risk
Disease-Modifying Anti-Rheumatic Drugs (DMARDs)
Class Indications: Rheumatoid Arthritis (RA), other autoimmune diseases.
Class Goal: Dampen immune activation, slow joint destruction (unlike NSAIDs which only treat symptoms).
Disease-Modifying Anti-Rheumatic Drugs
Doesn’t occur from joints, but from some flare up in joints
MOA: Treat RA by dampening innate and adaptive immune activation, block cytokine signaling, and altering synovial cell behavior reducing joint inflammation and destructive remodeling.
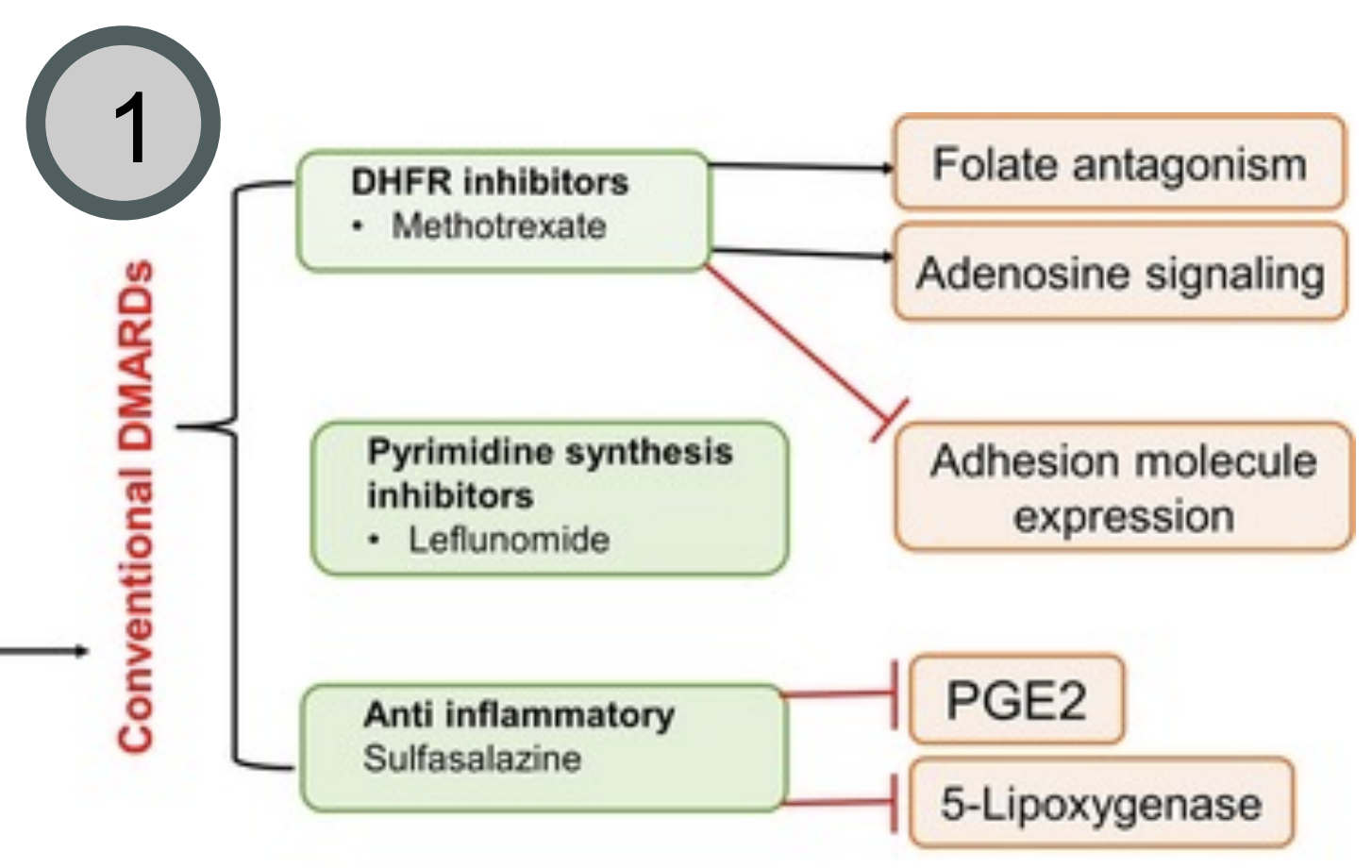
Convential Synthetic DMARDs
Agents that broadly modulate immune and inflammatory pathways associated with RA
Drugs: Methotrexate (main drug), Leflunomide, Sulfasalazine, Hydroxychloroquine
Used first line before or alongside other DMARDs and/or JAKis.
Methotrexate
Methotrexate
Class: Conventional Synthetic DMARDs
Main drug for RA & other autoimmune disorders
MOA: Inhibits DHFR & increases extracellular adenosine
Dose: Once weekly oral formulation (Class effect of DMARDs)
HAS AN ACTIVE METABOLITE
Co-administer folic acid to reduce GI toxicity/hepatotoxicity.
Methotrexate ADRs, CIs, BBW
CI: Non-neoplastic diseases during pregnancy
BBW: Serious ADRs and Embryo Toxicity (since inhibiting folate)
Immunosuppressive → risk of opportunistic infections + can affect live vaccines administration
DDI: NSAIDs reduce GFR (Increase drug levels) and antimicrobial drugs
Methotrexate (MTX) — Discovery & MOA
Origin
Developed in 1940s from folate metabolism research in cancer cells
Target pathway
Folate → needed for DNA synthesis (one-carbon transfer)
DHFR enzyme converts folate → active forms for DNA replication
Mechanism
MTX inhibits dihydrofolate reductase (DHFR) → DHF can’t be converted to active THF
~1000× stronger binding than folic acid
→ ↓ tetrahydrofolate → ↓ DNA synthesis (blocks rapidly dividing cells)
Clinical insight
High dose: anti-cancer (blocks tumor growth)
Low/intermittent dose: immunomodulation → ↓ inflammation in RA
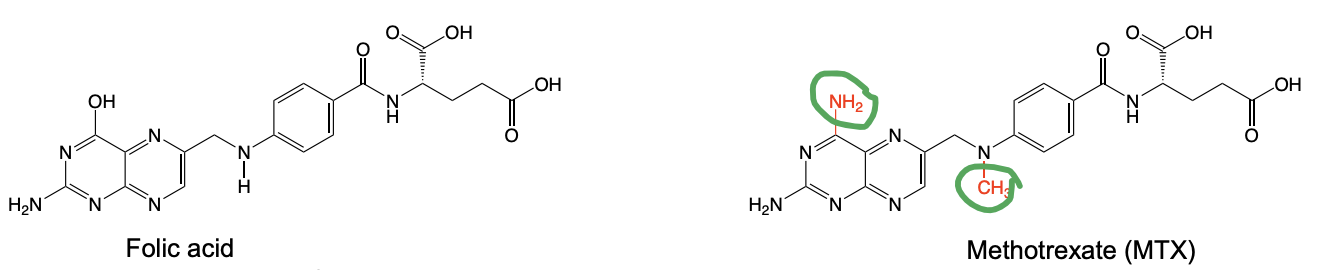
Folic Acid vs Methotrexate
Changes → increase potency of binding to DHFR
Considered an “anchor drug” for RA
Methotrexate MOAs
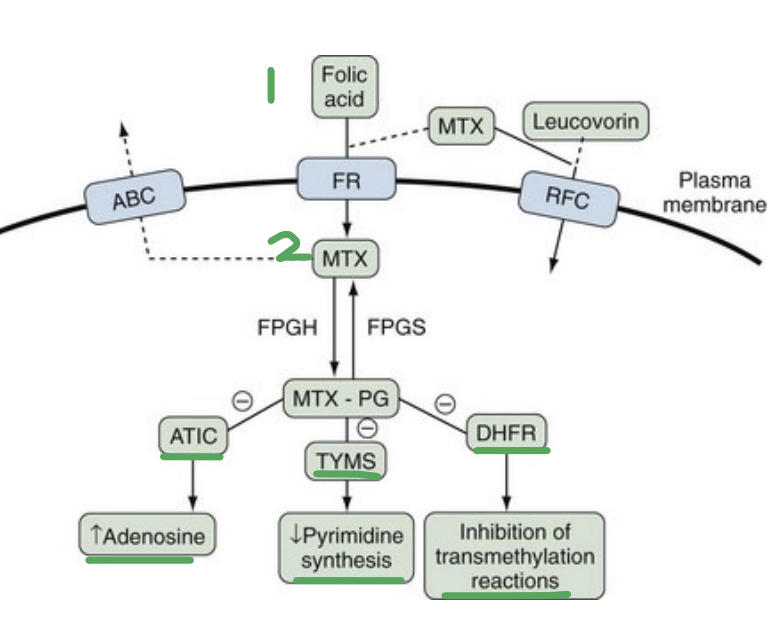
Enters the cell via Folate Carrier (FTC) and Folate Receptor (FC)
MTX → MTX-PG by foly-polyglutamtyl synthese to prevent it’s efflux by the ATP Bindinf Casseste transporter
Inhibits several enzymes (*KNOW*) →
Inhibits TYMPS (Thymidylate Synthetase) → Decrease pyrmidine synthesis.
Inhibits ATIC → Increases adenosine which has vasodilatory and anti-inflammatory effects.
Inhibits transmethylation of DNA, RNA, amino acids, etc needed for the cell to survive.
Leflunomide
Class: Conventional Synthetic DMARDs
Second line agent for RA (if fail Methotrexate)
MOA: Inhibits DHODH → decreases pyrimidine synthesis (1/3 of the MOA of Methotrexate)
Prodrug → Teriflunomide is the active form that exerts it’s effects.
ADRs (similar to Methotrexate): BBW for fetal toxicity, hepatotoxicity, DDI with hepatoxic agents.
Leflunomide
Sulfasalazine
Combined w/ Methotrexate to treat RA + ulcerative colitis
MOA: Metabolized in the small intestine to Sulfapyridine (SP) and 5-aminosalicylic acid (5-ASA).
5-ASA Effect: Inhibits PG synthesis
SP: Increases adenosine + Reduces pro-inflammatory cytokines.
ADR: No fetal toxicity (like Methotrexate and Leflunomide) but immunosuppression (class effect of conventional DMARDs).
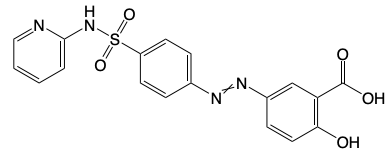
Sulfasalazine
Hydroxychloroquine
Class: Conventional Synthetic DMARDs
Add on agent to Methotrexate (like Sulfasalazine) for RA + Malaria
MOA: Increases the pH of T cells → blocks toll-like receptor activation and decreases cytokines
Racemic Mixture
ADRs: Bind to ion channels, effect anti-arrhythmic drugs, narrow TI drugs, cardiac effects and retinal effects (unique for this drug).
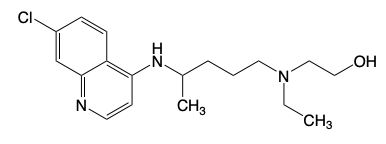
Hydrochrloquine
JAK Inhibitors
Drugs: Tofacitinib, Upadacitinib, Baricitinib (Suffix: -tinib)
MOA: Suppresses T-cell differentiation and proliferation, blocks B-cell signaling curbing pannus invasion, decreases TNF and IL-1 output.
Can have higher or lower efficacy than Methotrexate or TNF blockers but the trade off is long-term safety.
Black Box Warnings (vs. TNF blockers): Higher rate of death, MACE (heart attack/stroke), malignancy, thrombosis (PE/DVT).
JAK Inhibitors
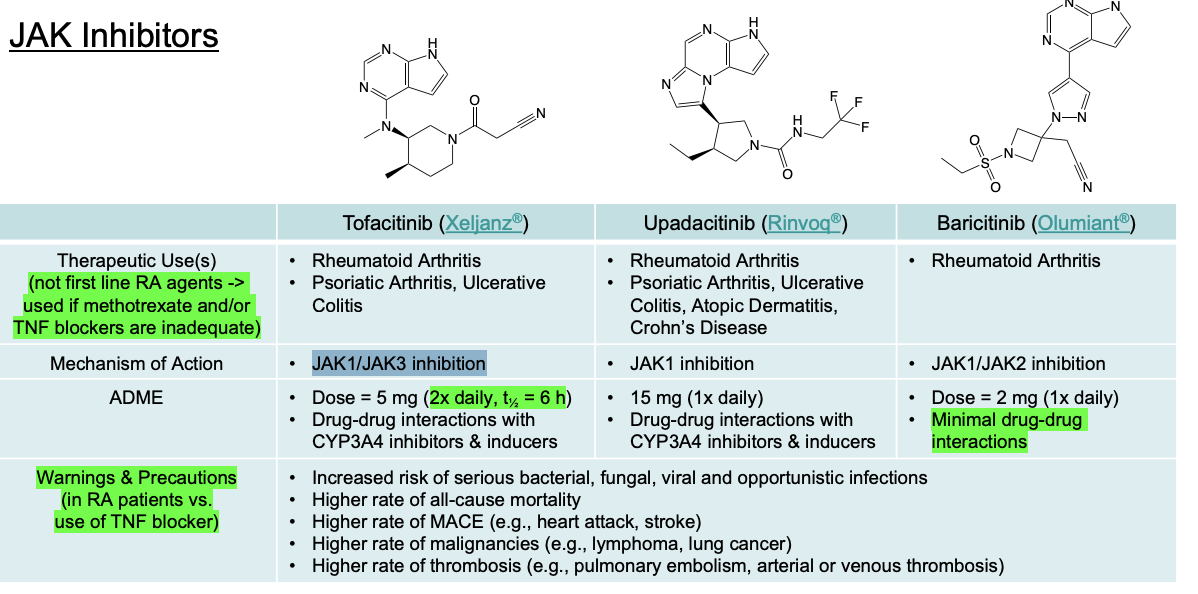
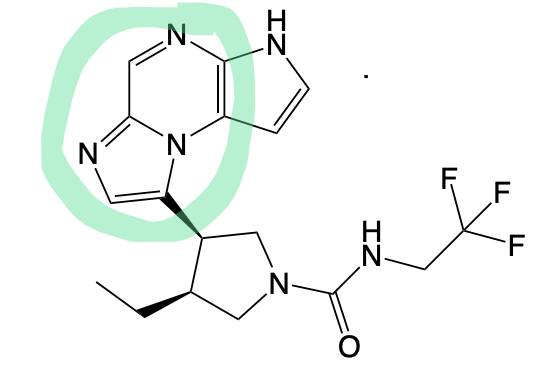
Upadacitinib (LOOKS LIKE A U)
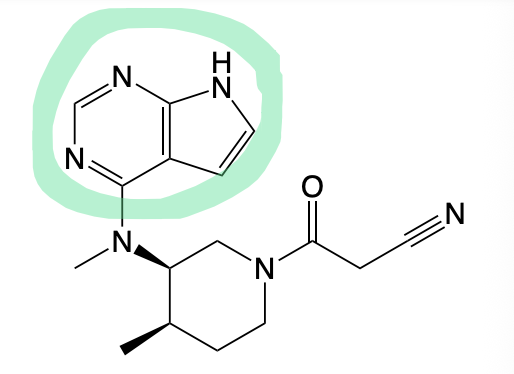
Tofacitinib
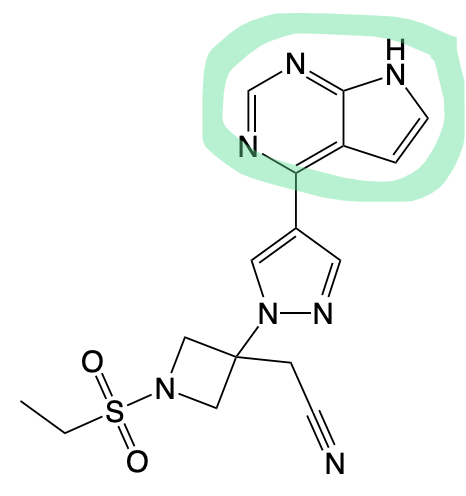
Baricitinib
Biologic DMARDs
MOA: Inhibit Tumor-Necrosis Factors (TNF) → TNF drives synovial inflammation & joint damage
Inhibition → Improves symptoms in RA (*synergistic w/ Methotrexate*)
ADR: Immunosuppression (DMARD Class Effect), increases CV risk and does enhance malignancy risk (but no meaningful difference).
Drugs: MABs
Biologic DMARDs (TNF Inhibitors)
Drugs: Infliximab (chimeric), Adalimumab (human), Golimumab (human), Etanercept (fusion protein), Certolizumab (PEGylated Fab).
Class Black Box: Serious infections (TB reactivation), malignancy (lymphoma).
Key Exception (Certolizumab): Lacks Fc region → no mTNF-cell cytotoxicity/apoptosis → may be safer in pregnancy.
Key Rule: MTX is required with Infliximab and Golimumab (recommended but not required for others).
MTX is required with ____ (TNF Inhibitors).
Infliximab and Golimumab
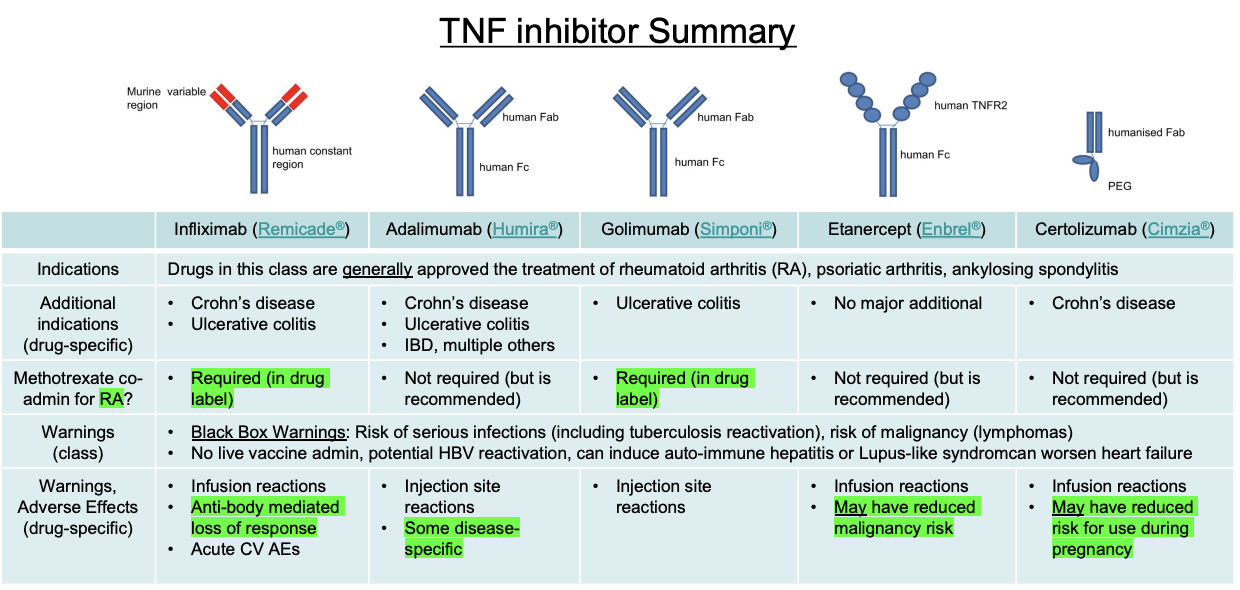
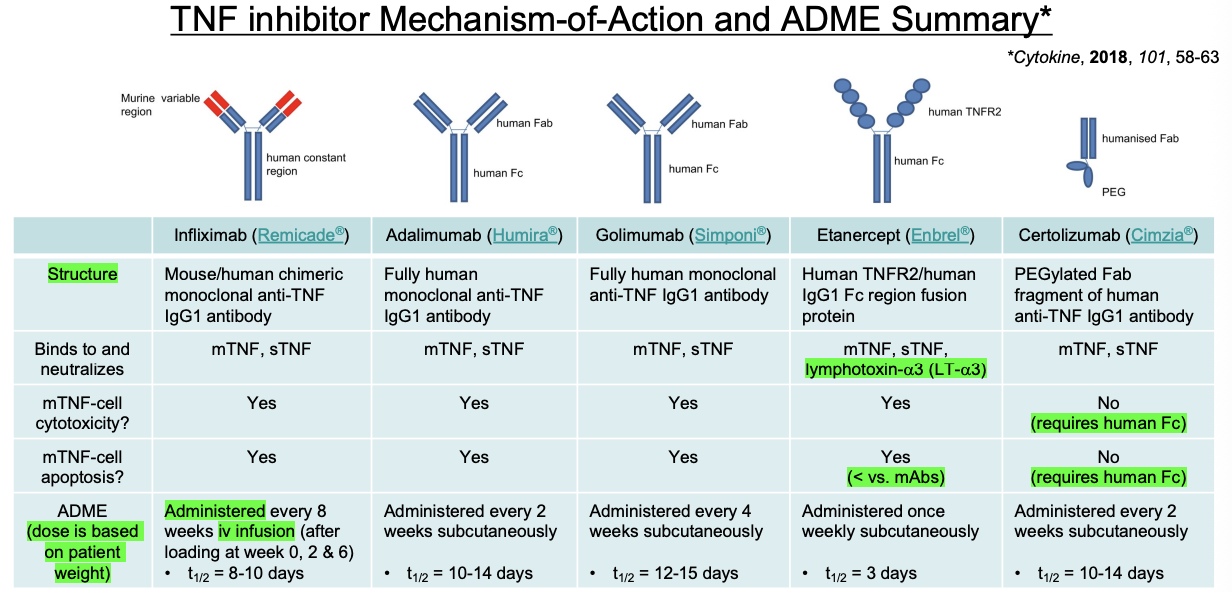
All TNF Inhibitors are SUBQ except…
Infliximab
Certolizumab has no ___
mTNF-cell Cytotoxicity or Apoptosis since this requires Fc Region