148 final (Set 4)
1/62
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
63 Terms
CAST
type V-K CRISPR associated transposon, targets but doesn’t cut so transposon can hop into a specific site. efficient and promising
targeted insertion of DNA is hard bc u need host cell repair machinery
CAST integrates DNA into target site at 80% success rate w NO selection
type V-K crispr-cas ALWAYS encoded within a tn7 like transposon
tns have their roles, tniQ the “communicator” between Cas9 and tns
Cas system only has tracrRNA and crRNA
kinda similar to dCas9-effector fusions
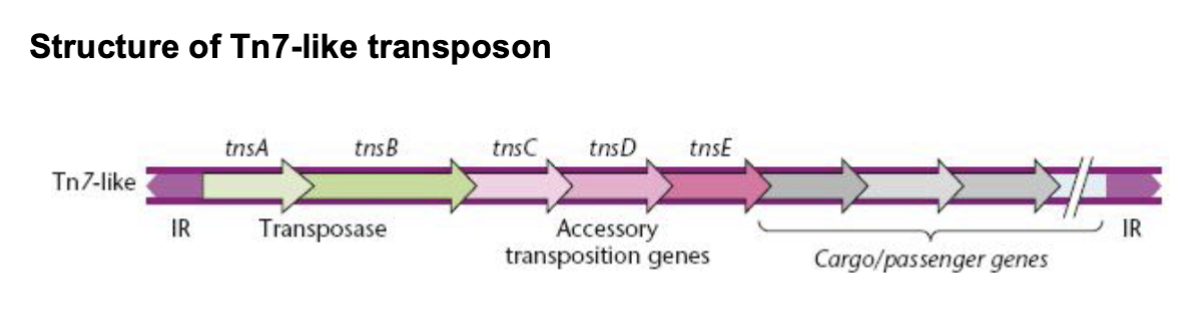
tn7 type transposon
has TnsA-E
tnsA/B = transposon
tnsCDE = accessory transposition genes
cargo genes next to them, flanked by IRs (RE/LE)
RNA guided DNA transposition / experiment example
TnsB/C and TniQ recognize IRs in Tn7 like transposon and excise it
tns/transposon complex binds to cas12K/crRNA/tracrRNA complex which targets transposon for insertion at site specified by crRNA
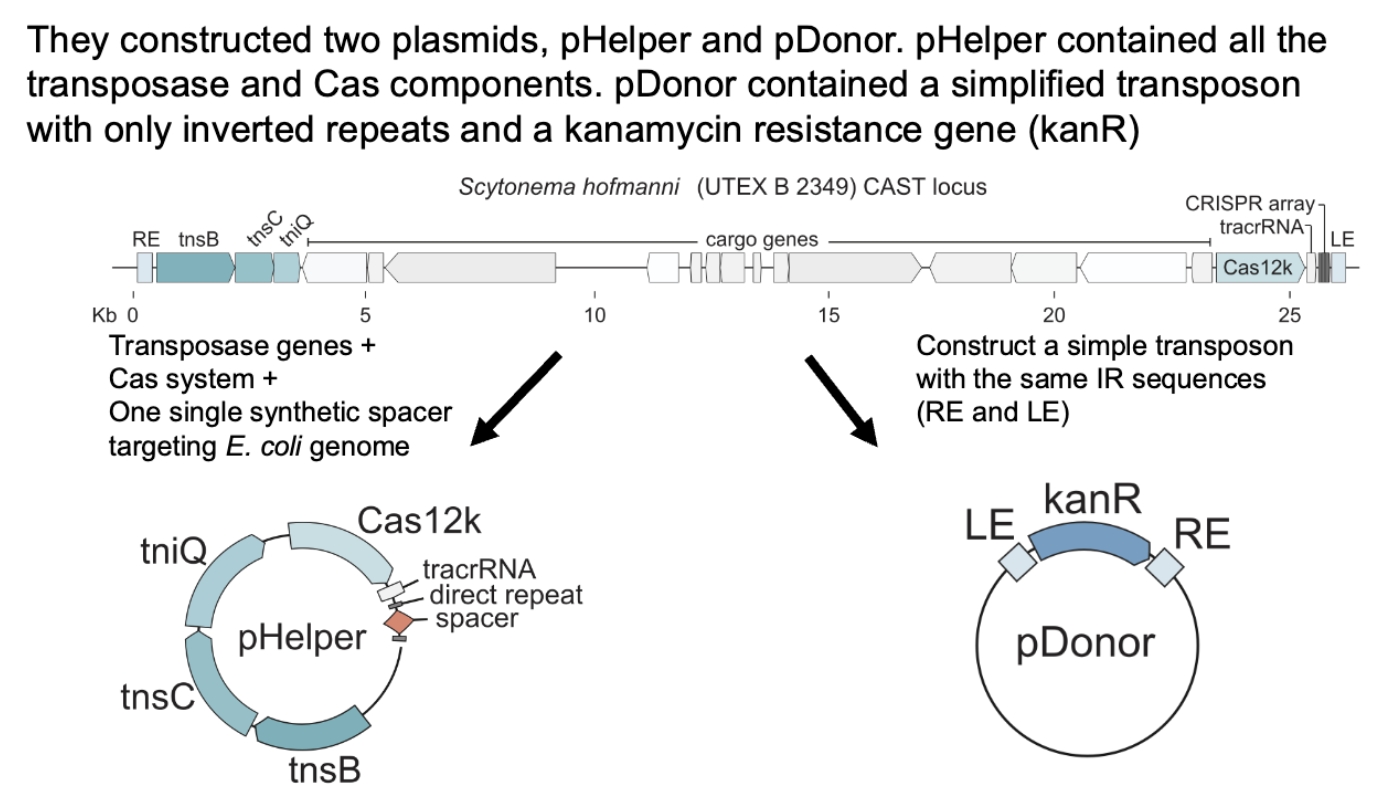
experiment:
pHelper has the crRNA, tnsB/C/TniQ
pDonor has just kanR and repeats
pHelper goes and snatches kan R through process above, expectation was that if cas12k and crRNA direct Tn insertion they’d see the kanR at the crRNA site.
CAST advantages
programmability of CRISPR + insertion capability of a transposon, don’t need to worry about homologous recombination machinery
transposition is more efficient + only requires transposase enzymes —> can happen in bacteria with low HR efficiency, doesn’t require a template homologous to target gene.
how we can determine which microbes are present in a certain envrionment
culture dependent (isolate microbe, study in lab)
culture independent (PCR based & 16s rRNA/metagenomics/transcriptomics)
16s rRNA profiling
why are microbes hard to culture sometimes
might have specific growth requirements
might grow super slow (esp relative to other microbes in sample)
bacteria might need presence of other organisms to grow
16s rRNA profiling
culture independent
9 variable regions and conserved regions
used to ID organisms
PCR amplify 16s gene using universal primers, compare to known 16s sequences, if they’re 97% similar —> same species
community analysis gives you:
species richness (distinct) & abundance (quantity)
Human Microbiome Project
goals
1) see how many microbial species there are in healthy adults using 16s profiling and metagenomics.
2) see dynamic changes that occur in microbiome of host under:
pregnancy/preterm birth, inflammatory bowel disease, stressors in folk w prediabetes
big pic: how does microbiome impact human health / disease, can we make a reference microbiome for typical individual
major conclusions:
there is NO REFERENCE MICROBIOME for healthy adults
major variability
one body site in individual is more similar to same site on someone else than different site on same person
tongue to someone elses tongue > tongue to same persons foot
Higher inter individual variation at species / strain level
ex: streptococcus main in oral, but theres a ton of types of them
GI Tract microbiome
GI tract has most diverse microbiome (gut has 10²/g, 300-500 species present), commensal relationship (they benefit, neutral for us)
colon is STRICTLY ANOXIC (no o2)
anoxia promotes growth of obligate anaerobes like clostridium (firmicute) and bacteroides (bacterioidetes)
^ ferment products of complex carbohydrates catabolism
phylum level diversity is low
big 3: firmicutes, bacteroidetes, proteobacteria
FB dominate in abundance
Roles:
develop early immune system
digestion
pathogen protection
influence how we process therapeutic drugs
factors about human microbiome that make research had

Auto-brewery syndrome / gut fermentation syndrome
consumed carbs are converted to alcohol by gut microbiota
candida / saccharomyces (fungal) & klebsiella pnumoniae (bacterial)
Bacteroides (GI microbiome)
obligate anaerobes fermentative species
Saccharolytic —> ferment sugars / proteins to acetate/succinate
bacteroides thetaiotaomicron:
prominent in large intestine
specializes in degradation of complex polysaccharides
increases diversity of plant polymers that can be degraded by human digestive tract
germ free mice
introduce specific microbes into them to see composition and effects
monocolonization, defined cocktail, complex
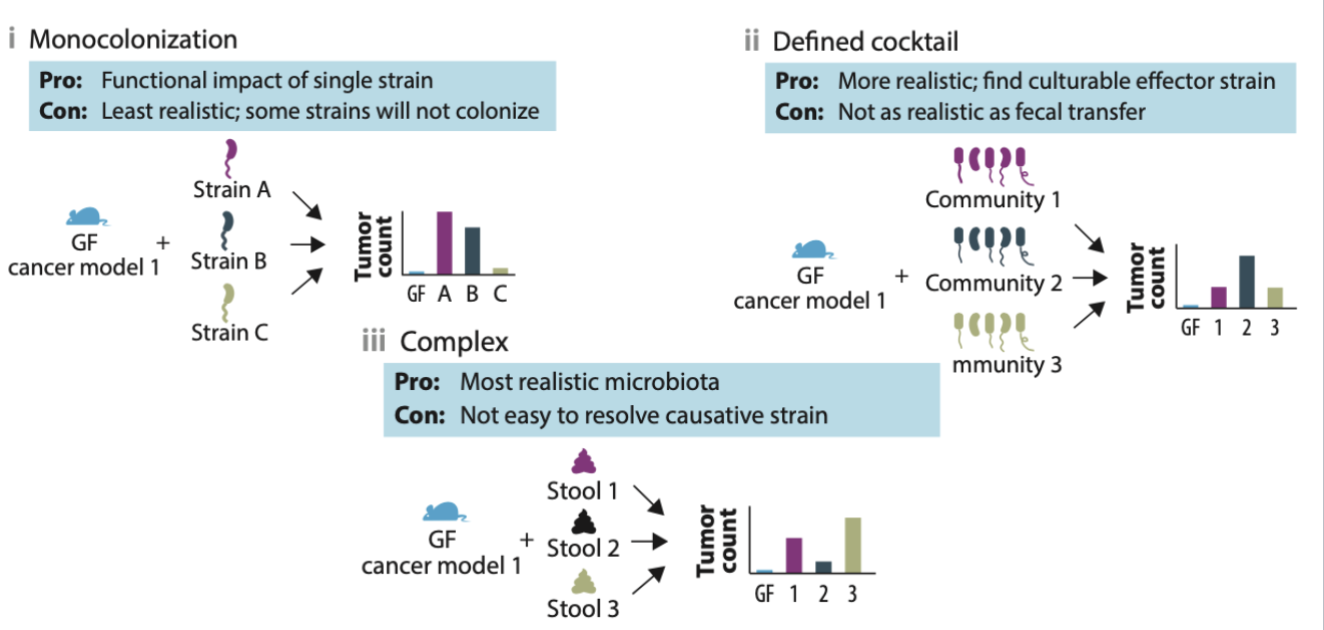
backhed et all (fat microbiome transfer)
gut microbiota is an important environmental factor affecting energy harvest from diet and energy storage in host.
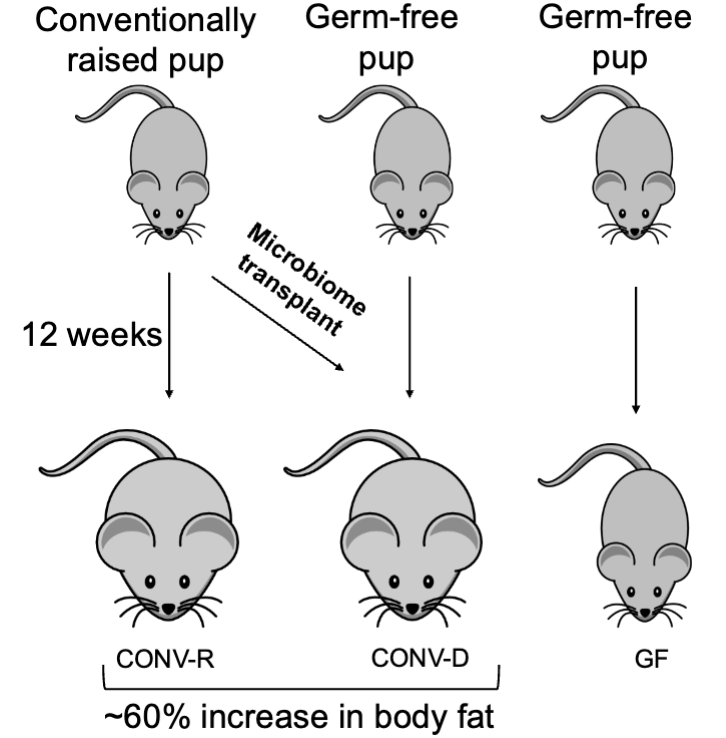
diet influences microbiome composition
western diet = high in fat w sucrose, malodextrin, corn syrup
^ cuts species diversity, body fat is higher
GF mice with western fed donor microbes vs cho donor microbes —> ate same amount but CHO donors only has 23%. body fat increase whereas 47% for western.
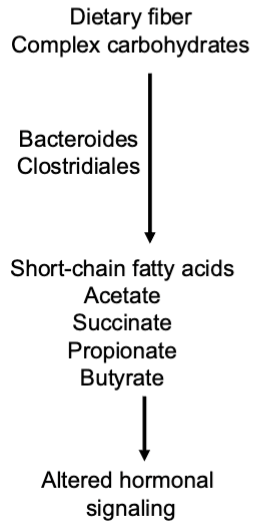
Challenges in native gut microbiome genetic analysis
1) delivery
DNA has to make it in efficiently without getting degraded by gut, other bacteria, chemicals, immune responses, washout
bc of all this, use conjugation
MetaEdit, megRNA decides where delivery happens
2) persistence of engineered bacteria
can be transferred then lost
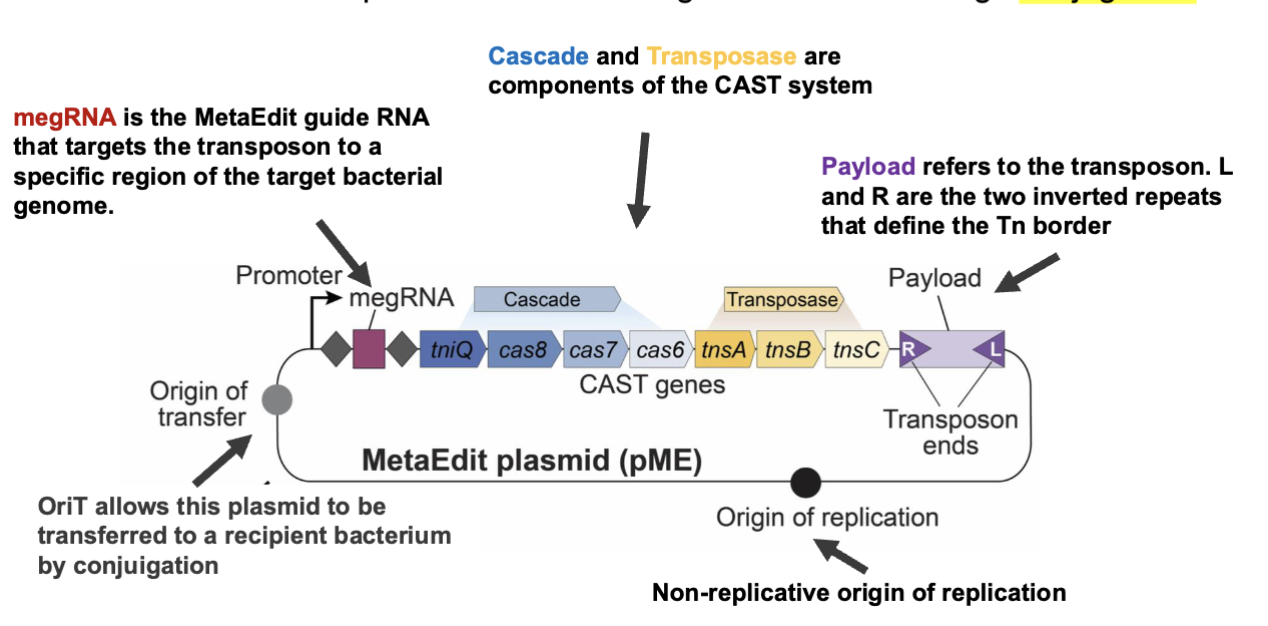
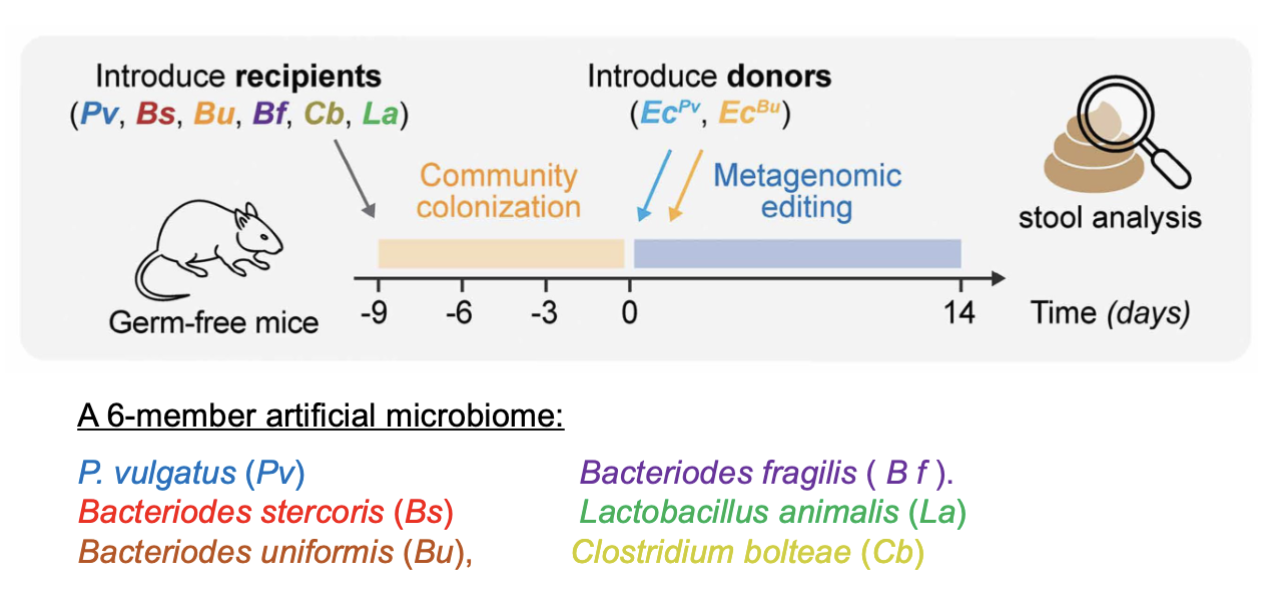
2 CAST experiments
GF mice given 6 commensal species (pic)
4% OV got the DNA precisely integrated but got lost in 10 days
they were only successsfully able to edit bacteroids thetaiotaomicron
selected for b thetawhatever that got the transposon by addiont 3 genes in transposon that give bacterium a growth advantage then gave them inulin (hella hard to digest, most cant)
BACTRINS (therapeutic e.coli donor strain that can be fed to mice infected w shiga toxin
BACTRINS uses CAST to deliver transposon into shiga toxin gene —> not virulent yay
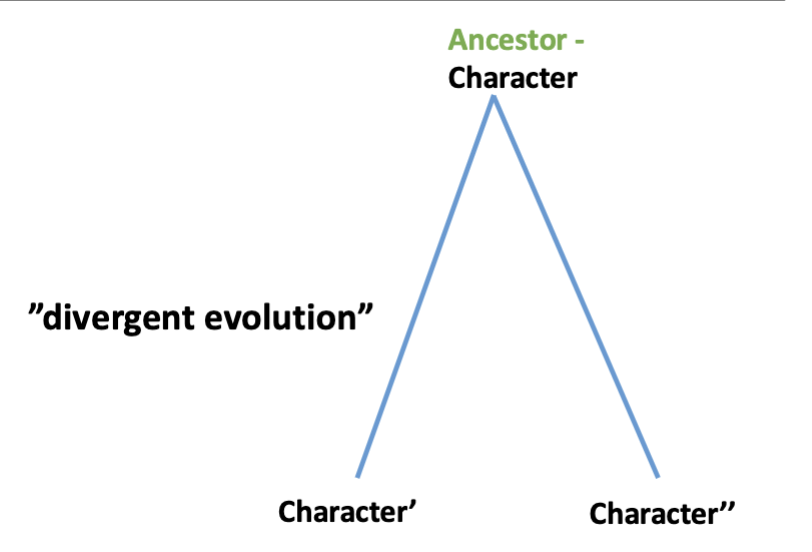
homology
similarity due to shared evolutionary history, common ancestral character
descended with divergence
bat/mouse forelimbs
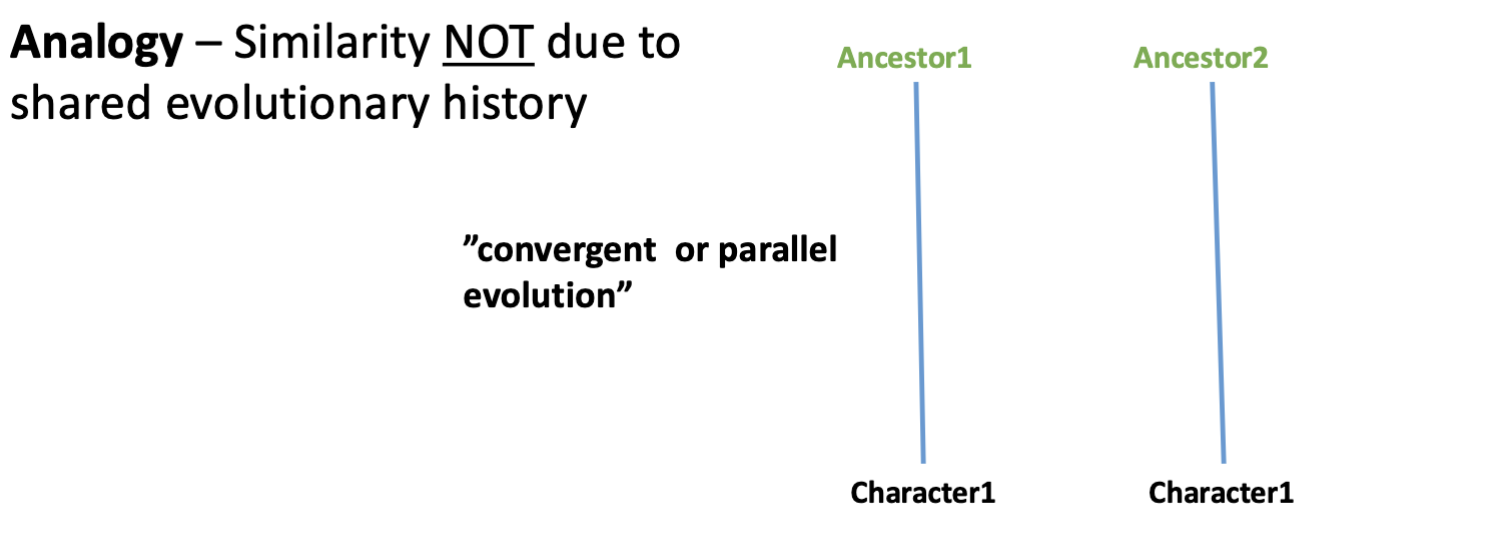
Analogy
relationship between 2 characters that descended convergently from unrelated ancestors
“parallel evolution”
birds and insects both evolving wings
but their evolution as forelimbs is homologous
gene/protein homology key concepts
1) G/Ps that have similar sequences are likely homologs but NOT ALWAYS (short sequences can be similar)
2) closely related homologs are likely to share some functionality but not always
enzymes that act on slightly diff substrates
3) in understanding function of uncharacterized protein, look for similar sequences, try to infer homology, can help predict function of protein
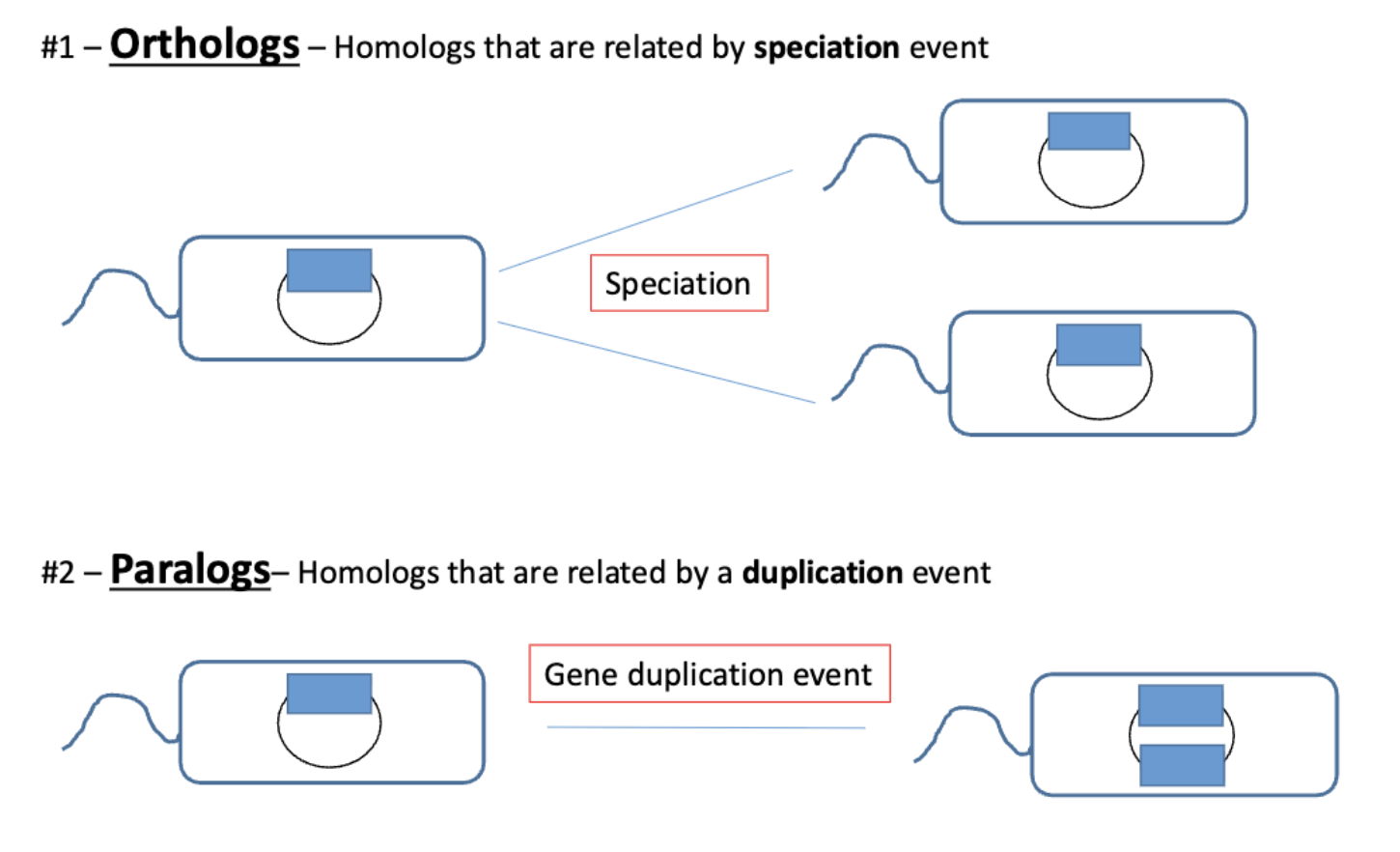
orthologs vs paralogs
orthologs are best for making species trees and good predictors of conserved function (shared functionality)
paralogs are likely to diverge in function
gene duplication is a major driver of genome evolution
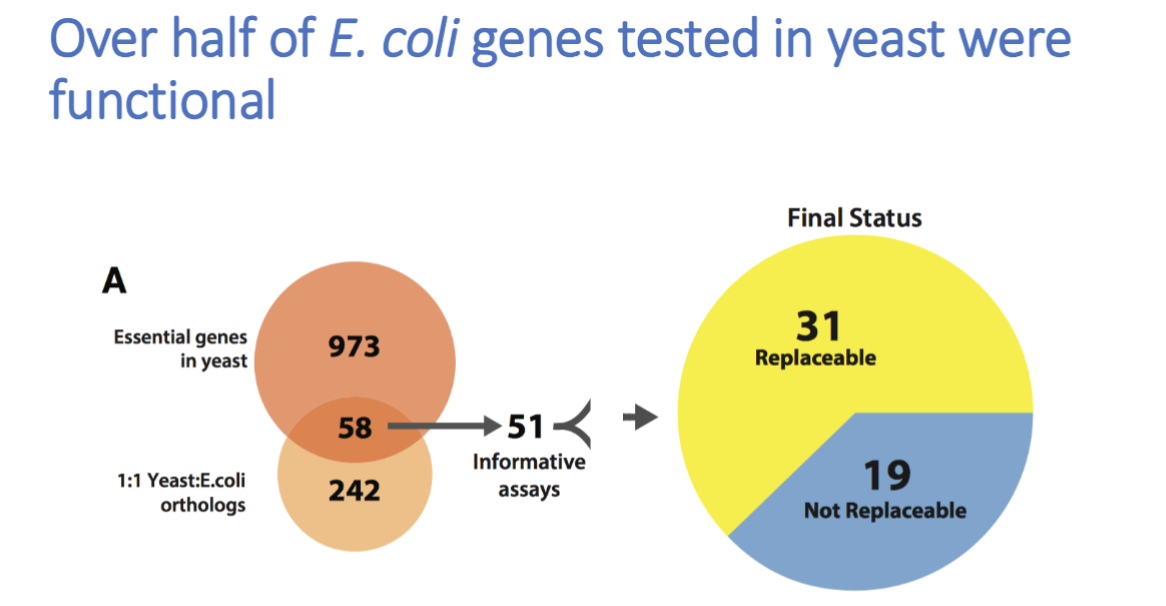
genetic complementation across domains (e.coli / yeast)
e.coli vs yeast example:
hella homologs, but functional equivalence?
experiment: rescue lethality of gene KOs in yeast by adding e.coli homolog
over half were functional, found that heme biosynthesis pathway is transferable from e.coli to yeast
Xenologs
relationship of any 2 homologs whose history since their common ancestor involves an interspecies (horizontal) transfer of genes
found when species and gene tree not matching
info u get from protein sequences
has 20 amino acids, order and composition = structure which = function
compare proteins to learn things boom sometimes more helpful than looking at whole protein

protein domain structural vs bioinformatics definition
structural:
protein domain = sequence that can fold independently and fulfill a minimal function
domain architecture = arrangement of domains in a protein
protein complex = group of proteins that come together to perform a biological function
bioinformatics:
protein domain: sequence matching a statistical model that corresponds to known or predicted domain
domain architecture: set of domains comprising a protein
protein complexes: not directly genome encoded
protein annotation vs domain annotation
protein:
find full length orthologs among proteins of known function
danger —> overannotation and assuming ohmology = shared function
domain:
finding which known protein domains are present in the sequence
Protein function prediction reasons?
1) work with protein sequence bc DNA evolves faster than protein
2) groups of amino acids have similar chemical properties, we can compare proteins by similarity not just identity
BLAST
heuristic approach, approximates smith waterman algorithm for local sequence alignment
guaranteed to find optimal local alignment between 2 sequences, but BLAST isn’t
but its way faster!
bit score = log2 scaled and normalized raw score
higher bit score = higher similarity
Expected value (e-value) = # of hits of equal or better quality that could be found by chance
lower values more significant
varies based on size of database you’re looking at
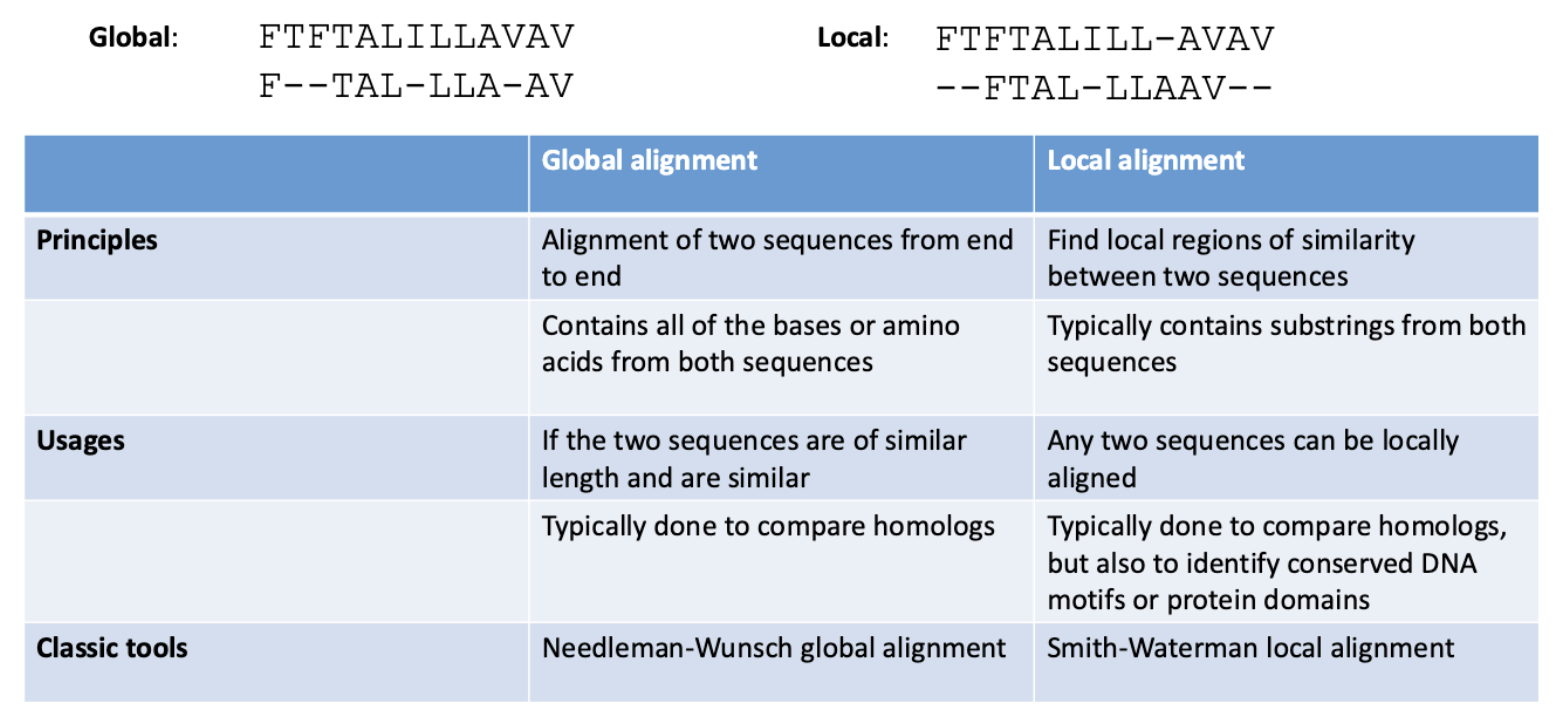
global vs local alignment
protein BLAST problems
limited at finding distant homology
doesnt take into account conservation / functional importance of individual amino acids
based on local similarity, can lead to annotation errors & propagation
BLOSUM62
amino acid substitution matrix (pBLAST default)
compares amino acid substitutions among proteins no more than 62% identical
sees if swaps are significant or not
can help find homologs of moderate distance (62%)
Profile search methods
take into account conservation of individual amino acids residues in a group of homologs, calculate position specific scoring matrices (PSSMs)
PSSMs useful for IDing more distant homologs
tools: PSI-BLAST, Profile hidden markov models (PFam)
Multiple sequence alignment
used to calculate a statistical model that corresponds to a known domain or predicted domain
PSSM: for every position in alignment, make custom scoring matrix
conserved positions: high positive score for conserved AA, negative for not conserved AA
Annotating protein domains (Pfam and InterPro)
Pfam = collection of statistical models of protein domains
InterPro = meta collection of protein models including multi-domain ones
protein domain annotation can be used to gather homologs
Pfam answers "does my protein contain domain X?" and InterPro answers "what is my protein, based on everything we know about all its parts?
AlphaFold
predicts lowest energy folding state for protein from structure alone
uses the fact that residues in proximity evolve together + 3d info
Two proteins can have completely different sequences but still fold into the same 3D shape —> reveals they're related in ways sequence comparison alone would miss.
Protein language models
sequence diversity analysis
databases learn protein sequence patterns then can use that knowledge to predict sequences
Metagenomics
direct genetic analysis of genomes contained within an environmental sample
culture independent approach
what is there, what are they doing
2 types:
Marker gene (amplicon)
PCR amplify and sequence a conserved marker gene from community to see what is there
deeper in on one sample, cheaper and faster
genome-resolved (shotgun)
sequence everything, can use larger samples
marker gene vs amplicon

Marker-based / amplicon metagenomics
Amplicon-based “metagenomics” is useful for “What is there?” questions; and for “How does the microbial community change in response to …....?” question
1) ID microorganisms that have a conserved marker gene —> PCR amplify it
^ primers don’t always catch everybody tho
2) sequence using illumina / NGS
3) compare to known database
commonly used marker genes:
16s rRNA (bacteria / archaea)
ITS (fungi)
18s rRNA gene (euks)
COX1 (animals)
single copy highly conserved protein coding genes

16s RNA / ITS
prokaryotes:
conserved/variable, interacts with RBS, vertically inherited, in all prokaryotes
use universal primers to amplify
eukaryotes:
not enough variable regions to do 16s —> ITS instead
iTS primers use conserved regions in rRNA genes to amplify non-coding regions between them
16s rRNA based marker gene approach to characterize microbiomes
advantages:
cheap and fast
readily done even with low amounts of DNA (bc PCR)
hella samples can be processed/multiplexed (good for temporal studies / large surveys of community structure)
disadvantages:
link between 16s rRNA sequence and functional capacity of microbe isn’t great (don’t have genome)
many sources of bias (DNA not represented or chimeric)
16s primers work on most but not all bacteria
16s profiling experimental setup
1) isolate DNA from all environmental samples
2) PCR amplify 16s rRNA genes frome each community, sequence amplicons w/ illumina
bruh what
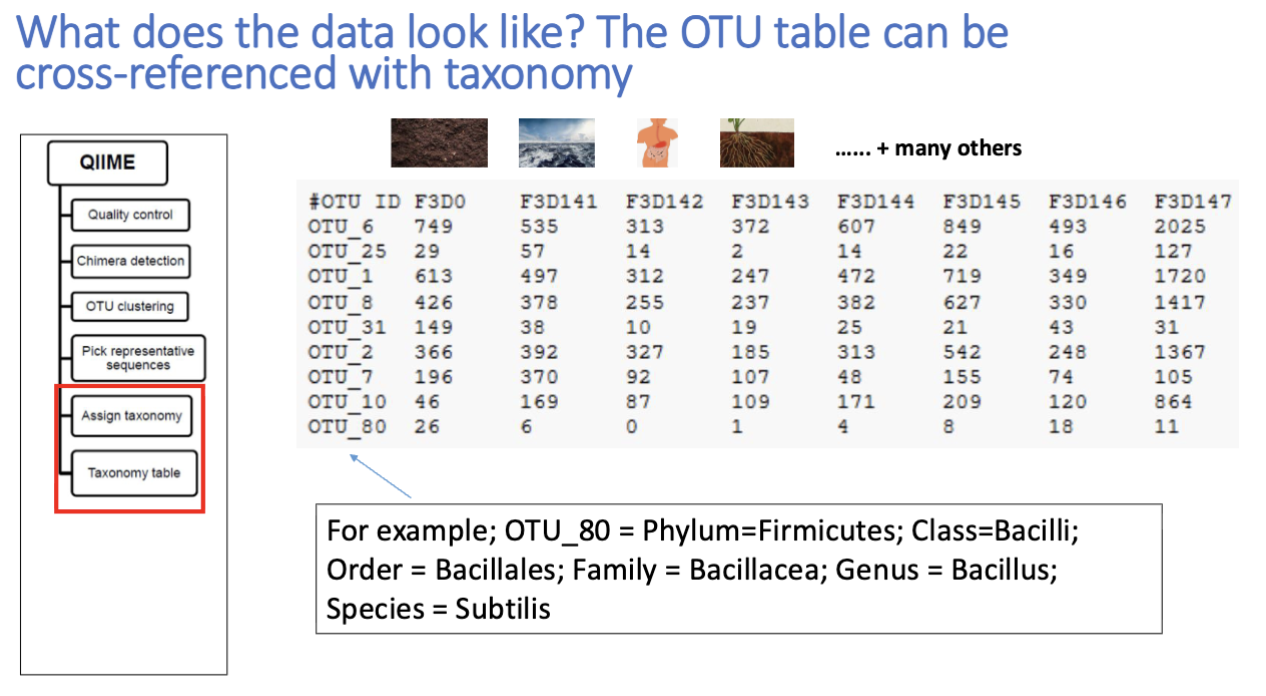
operational taxonomic units (OTU)
unit to classify groups of similar organisms
clustering performed on a group of sequences
OTUs are defined by user (operational)
ex: 97% identity of 16s rDNA gene for OTU is human made stat
OTU clustering: grouping together closely related sequencing reads from a sample
OTU is just sequences you think are the same, then cross referencing actually identifies it
higher resolution attempts to make the threshold higher than 97%
amplicon sequence variants (ASVs)
exact sequence variants (ESVs)
zero radius OTUs (zOTUs)
Earth Microbiome Project
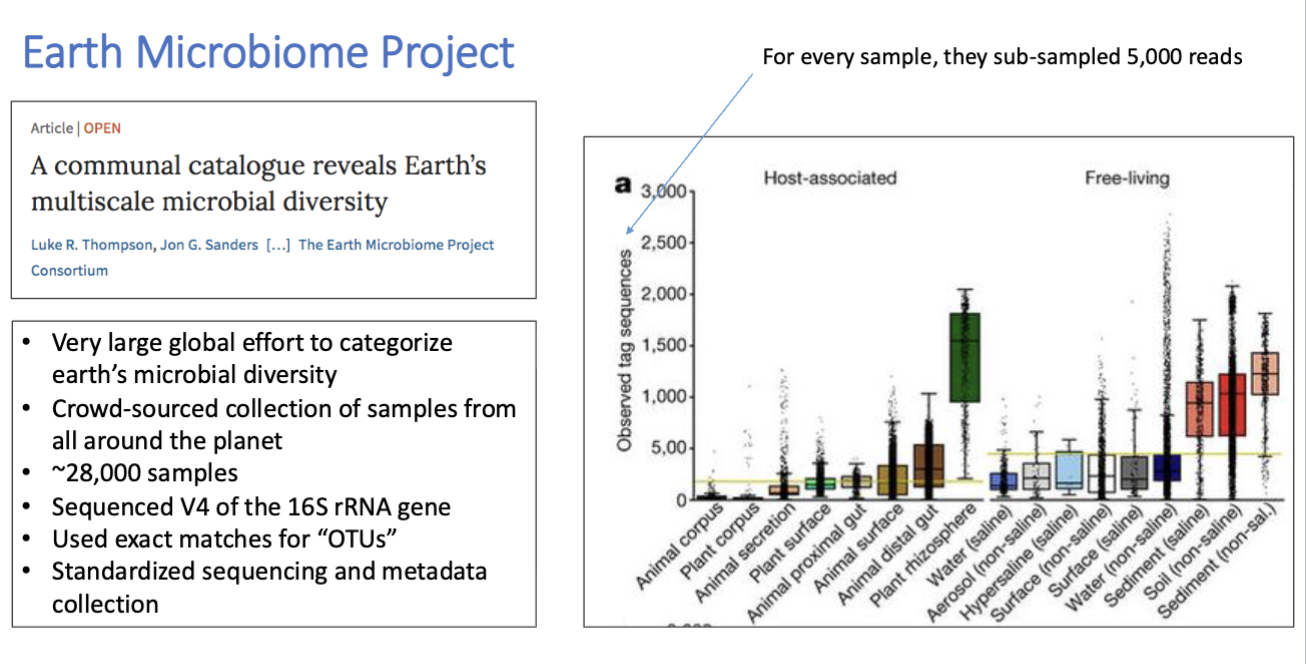
What can 16s rRNA based metagenomics tell you
microbial abundance profiles at diff taxonomic levels
alpha diversity measures micorbial community composition
shannon index auuuughhghhghghghgh
measures for a single sample
richness and evenness
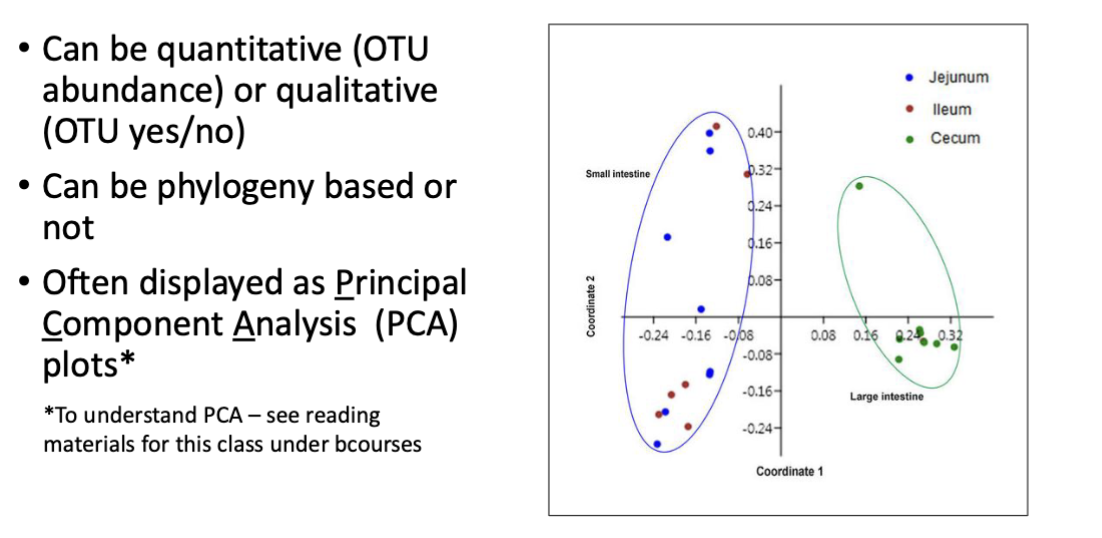
Beta diversity: measures microbial community differences
quantitative (otu abundance, same organisms in same amount?)
qualitative (otu yes/no, share same organisms?
displayed as PCA (principal component analysis plots) pic
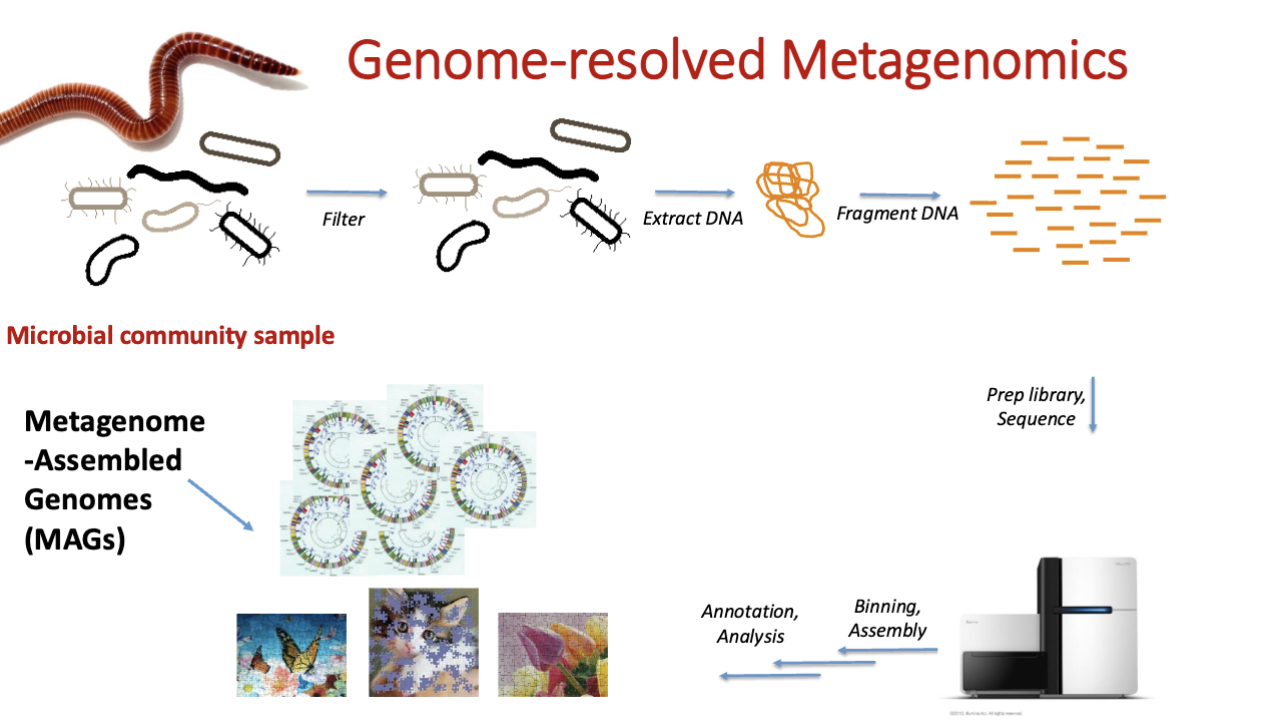
assembling whole genomes from metagenome challenges

Binning in metagenome sequence assembly
can be based on read abundances, GC content, tetranucleotide frequency
2 types:
1) bin reads then assemble: bin reads, assemble into contigs, genomes
puzzle pieces together then make the picture
2) assemble into contigs, then bin
put pieces together in squares, then put them together to make the picture

metagenome assembled genomes (MAGs)
what u get after the binning, “draft genome”
annotation of MAGs
1) structural, ID features in genome
2) functional, assign putative functions to features via sequence similarity, homology, protein domains, predicted protein structures
assessing quality of MAGs:
in a MAG, look for single copy genes (SGCs) that are universally present in related bacteria
% completeness: estimated by % of marker genes present in MAGs
contamination = estimated by # of instances of more than one copy in marker gene
high quality MAG has >90% completeness and <10% contamination
SCGs used to assess genome completion/contamination oare orthologs & homologs
expanded tree of life (CPR)
Candidate Phyla Radiation (CPR)
bacteria (many novel phyla):
• small genomes (~1,000 genes)
• small cells
• Incomplete biosynthetic
capabilities (ex. Amino acids)
• May be obligate symbionts or
parasites of other organisms
genome resolved metagenomics summary

Functional Metagenomics
microbes r so important but we cant culture all of them
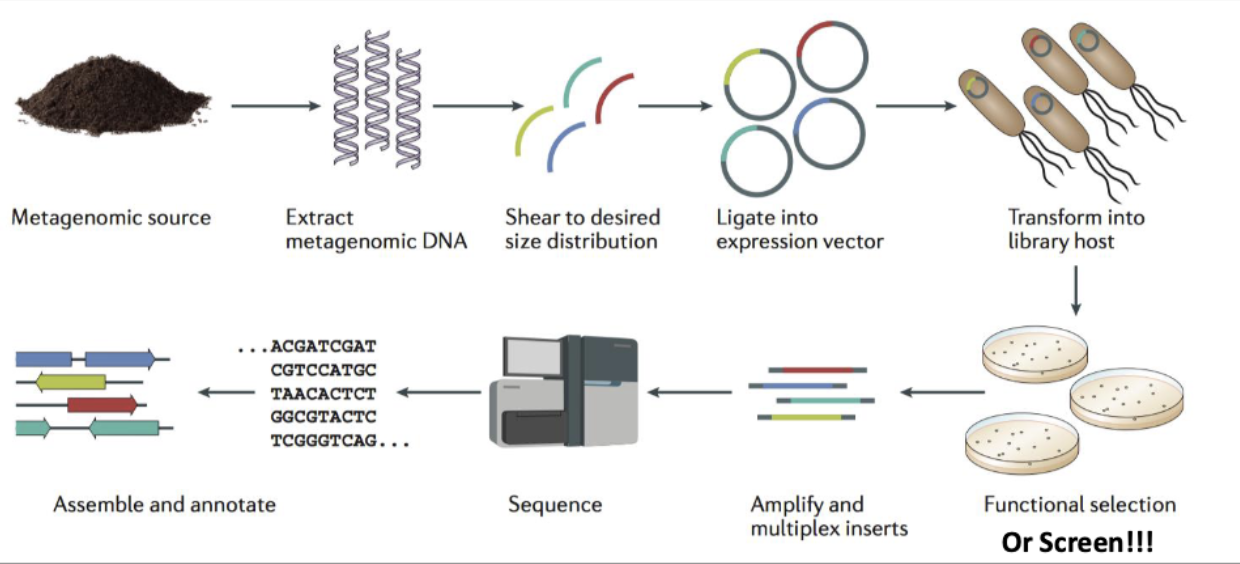
Functional metagenomics is the identification of new compounds or genes with desired activities from environmental DNA
need to express DNA in e.coli (heterologous host)
new gene functions from metagenomic DNA can be IDd by selections (of e.coli) or screens
selections: antibiotic resistance genes
screens: new phosphates
discovery of new antibiotics from metagenomic DNA
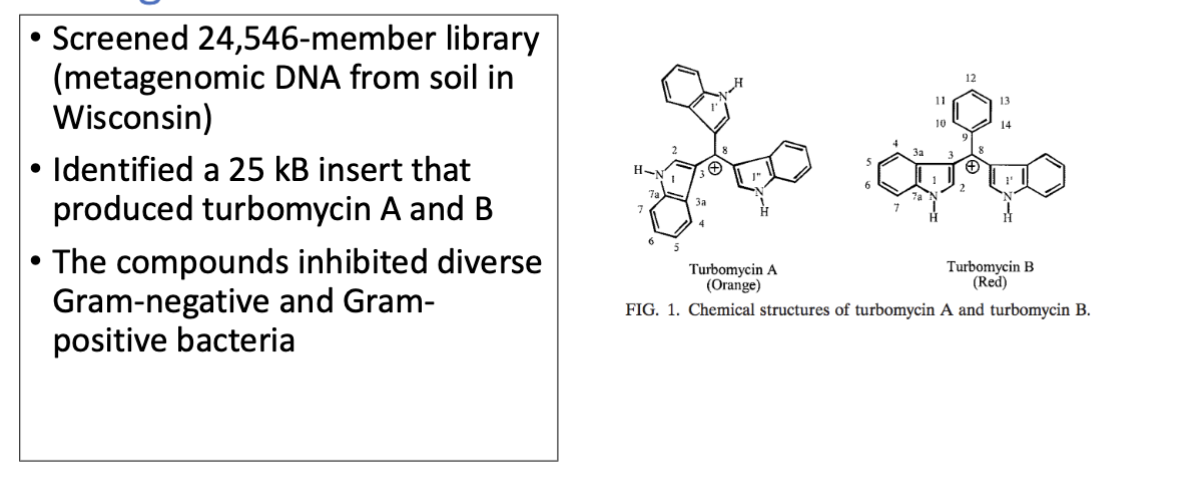
IDing new functionally tested genes from metagenomic DNA
spread of antibiotic resistance
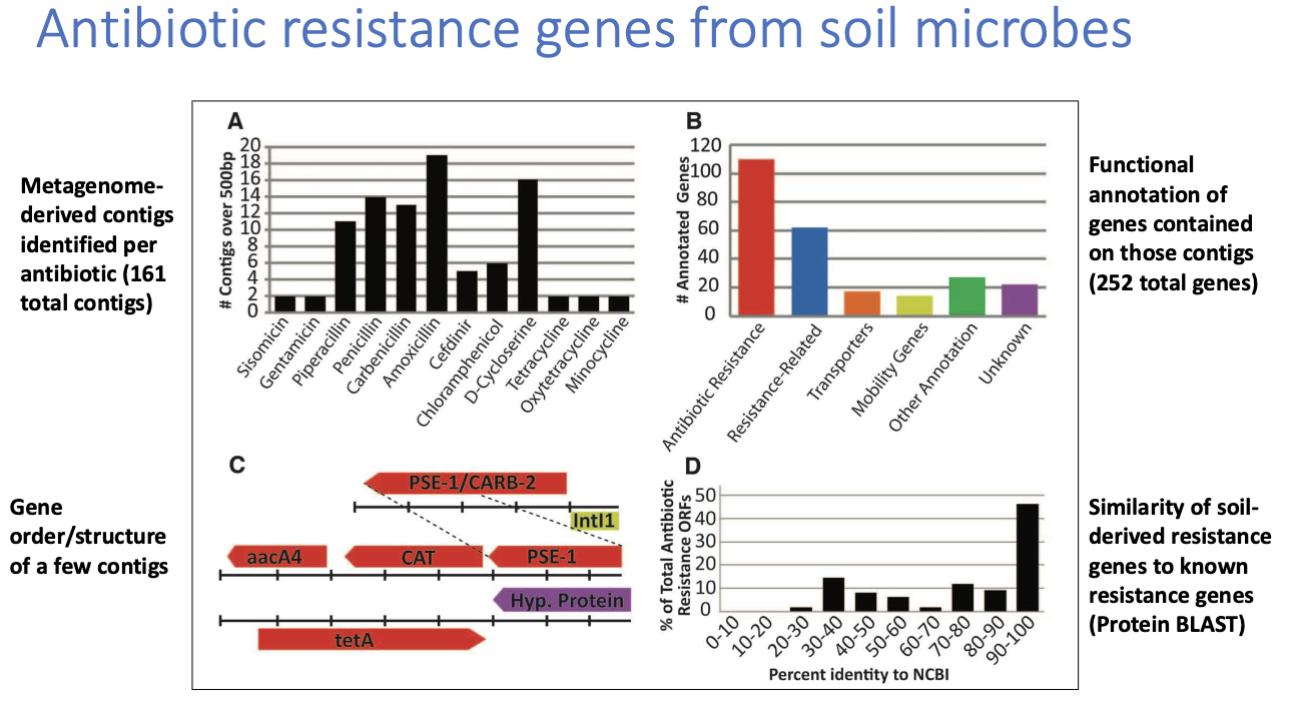
Func genomic application 1: identifying antibiotic resistance genes from natural envrionments
q: Have pathogens and soil bacteria recently exchanged resistance genes (if yes then sequences should be similar)
approach: func metagenomic using DNA soil from microbes
found that some resistance genes from soil were 100% identical at nucleotide level to genes from pathogens
evidence of HGT of resistance genes between soil bacteria / pathogens found
Func genomic application 2:
q: are novel phosphatases that escaped detection
approach: func metagenomics using DNA from soil microbes
experimental approach + Add an indicator compound to agar plates that makes phosphatase-positive colonies a different color
BCIP turns phosphatase including ones blue
21 e.coli colonies with phosphatase activity found —> next step BLAST to gather putative homologs, protein domain annotation, structure modeling.
find proteins with similar sequences

common functional metagenomics challenges

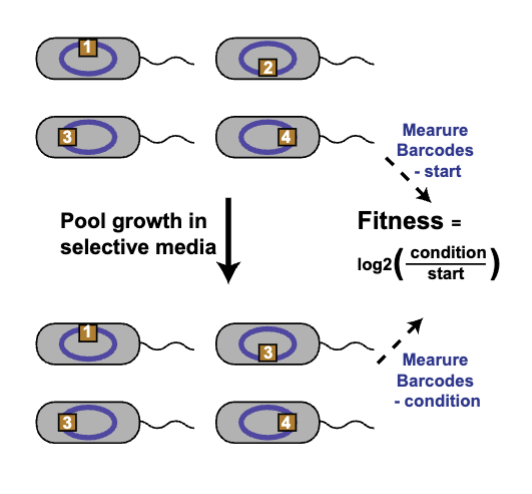
Tn-Seq
forward genomics tool
importance of thousands of genes assayed simultaneously in one pot
all tn strains grown together in same culture and compete
important genes for fitness IDed through NGS
genes providing benefits under certain conditions get DEPLETED during tn seq
Essential genes
required for growth
conserved across species (usually)
10-20% genes are essential typically
CRISPRi for functional genomics
The mechanism:
You design a guide RNA targeting the promoter or early coding region of a gene you want to silence
dCas9 + guide RNA bind that location
dCas9 just sits there — physically blocking RNA polymerase from transcribing the gene
No transcription = no mRNA = no protein = gene effectively silenced
repressor domain called KRAB in euks
CRISPR cas in bacteria
1) adaptive immune system in prokarotes:
infection, bacteria gives phage protospacer to CRISPR array
CRISPR array transcribed into RNA
RNA —> crRNA (1 per spacer) —> cas + crRNA
cas patrols cell using this, if it recognizes dna match, creates a ds break in phage DNA —> phage is done for
2) tracrRNA + crRNA —> sgRNA + Cas = systsm with 2 parts that can cut at a specific location in a genome
targeted mutation via ds break at a specific sequence
BUT NHEJ lethal in bacteria
3) community editing of microbes using CRISPR guided transposons
CAST to guide where they go?
Diff functional genomics approaches based on CRISPR
1) ds break —> loss of function mutation in target gene
2) fusion of transcriptional activator = overexpression of target gene
3) fusion of transcriptional repression = repression of target gene
performing genetics research on uncultivated microbes