cancer 2
1/24
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
25 Terms
describe the MAP pathway
growth factor binds tyrosine kinase
GEF activated so GDP -> GTP
RAF phosphorylated
MEK phosphorylated
ERK phosphorylated
transcription factors phosphorylated
genes involved in cell division activated
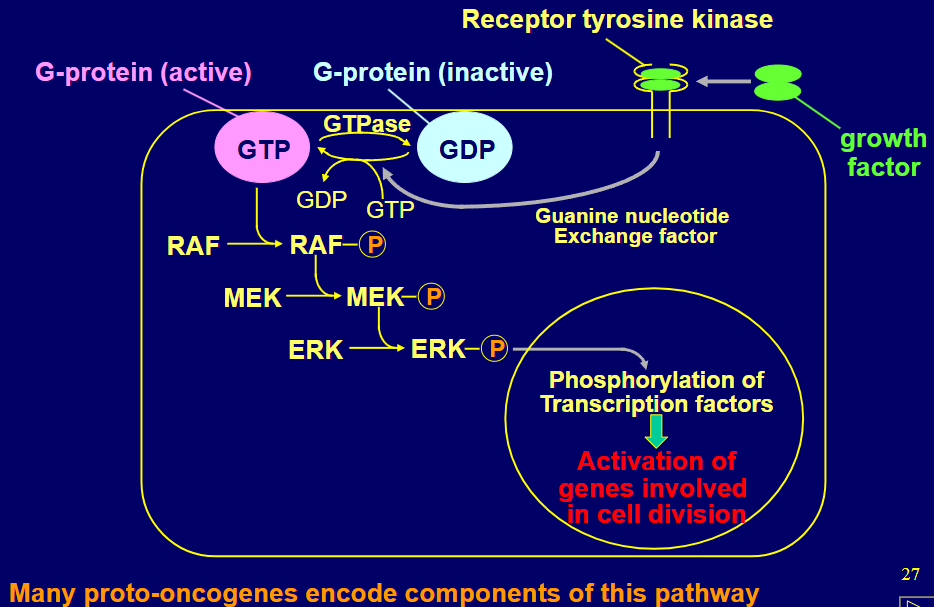
give some examples of proto oncogenes in the MAP pathway (activation of any allows cell division in absence of growth promoting signals)
FGF3 & PDGFB (growth factor, ligands)
BRAF & MEK (cytoplasmic kinases)
MYC & MYCN & FOS (transcription factors)
describe the Myc example
regulates may cellular functions including growth, DNA repair & apoptosis, upregulated by APC mutatnts
Myc deregulated, overexpressed via retroviral insertion/ chromosomal translocation next to Ig promoter, gene amplification, deregulated cell signalling (Myc regulate 10-15% transcriptome)
describe the bcl mutant
overexpressed B-cell =resistant to cell death - didn’t die as much when grown factor F removed, didn’t die at all when Myc & B-cell overexpreesed (mimicked transformation)
B-cell2 inhibits apoptosis by binding Bax/ Bak/Bok on mitochondria
usually BH3-only proteins inhibit Bcl2 under stress to release this & activate apoptosis
what happens on B cell leukaemia
Bcl2 translocated so transcription regulated by Ab heavy chain enhancer -> overexpressed -> enable B cells to resist apoptosis so numbs build up in blood -> leukaemia
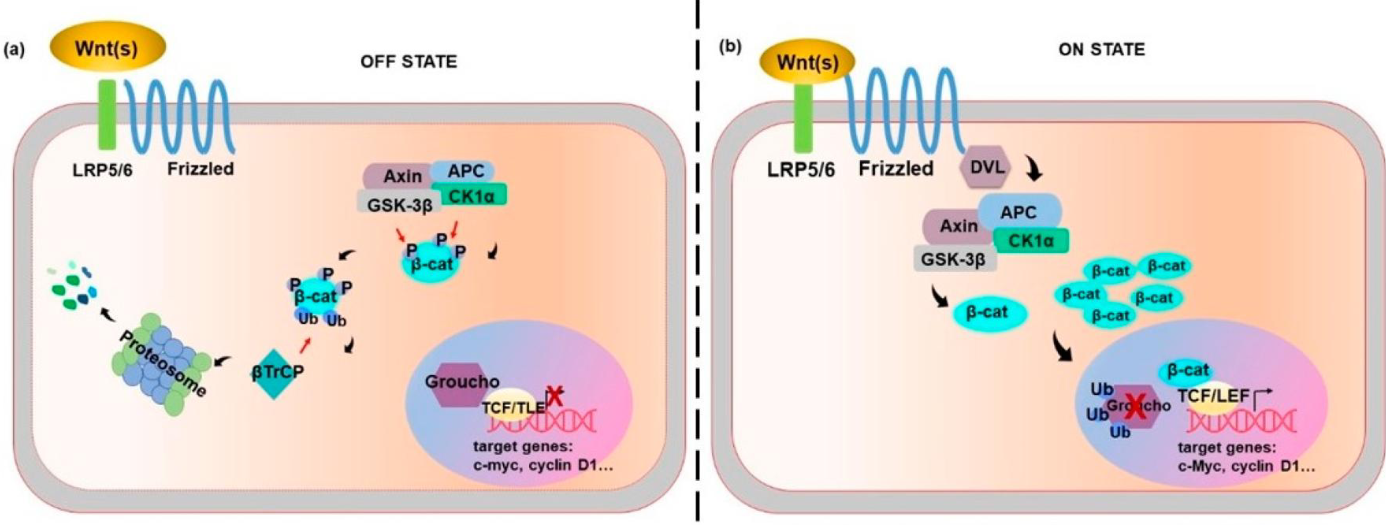
describe the Wnt pathway
Wnt binds frizzled receptor -> B-catenin no longer phosphorylated & ubiquitinylated (not broken down by proteasome) -> binds target genes (C-Myc, cyclin D1) & Groucho is ubiquitinylated instead (Groucho inhibits TCF/TLE transcription)
what are tumour suppressor genes usually
inhibitors of cell cycle progression, proteins needed to sense/ repair damage to DNA (e.g. Tp53 & APC)
describe familial adenomatous polyposis
inherited defect (causes truncation) in APC (adenomatous polyposis coli), autosomal dominant, acused by mutation in germline cells in APC gene on chromosome 5q2
loss of heterozygosity on normal chromosome 5 -> FAP (in children, when normally seen in 50+ years from sporadic loss of both)
APC is part of complex that phosphorylates beta catenin so w/o it beta catenin activates TCF & LEF
(can also cause desmoid tumours, osteoma, adrenal adenoma etc.)
describe the function of tp53
encodes p53, usually recessive, usually degraded rapidly
but if damaged DNA present → phosphorylation of p53 increases its stability
p53 _ activates gens CDKN1A (p21) inhibitor of cyclin-Cdk complex needed for cell cycle progression (halting at G1-S boundary until repair DNA), if badly damaged DNA -> BAX stimulates apoptosis
mutation -> damaged cell no cell cycle arrest -> increases mutation -> genomic instability (damage cell doesn’t undergo apoptosis)
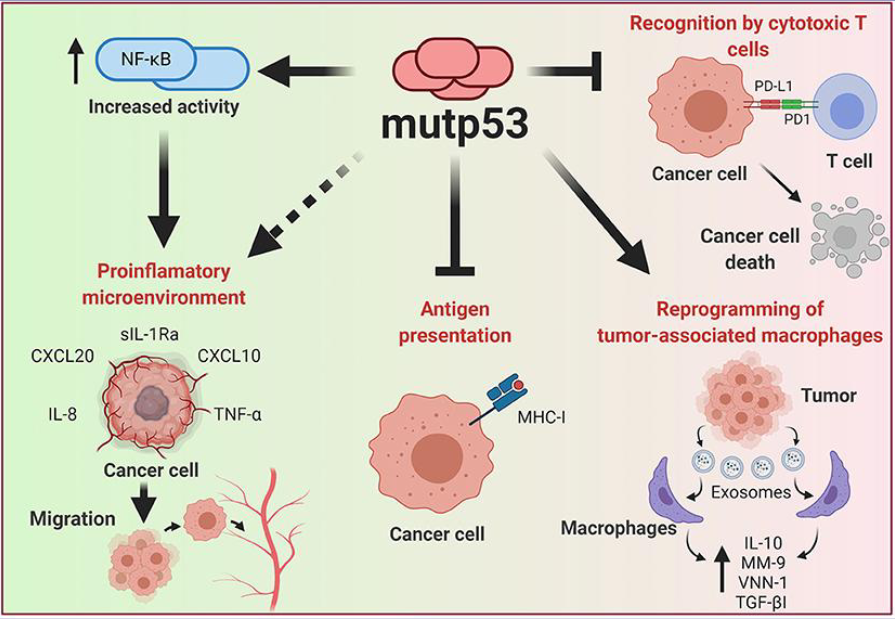
what can gain of function mutations in tp53 (in the DNA binding domain/ protein folding region)
reprogram macrophages to be pro tumorigenic
PD-L1 levels increase -> blocks T cell killing
increasing activity of Nf-KB to increase inflammation
increases migration
inhibit antigen presentation
give 2 example of tumour suppressor gene mutations
CDKN2A (cell cycle inhibitor) LOF -> melanoma, pancreatic cancer
BRCA1 & BRCA2 (involved in DNA repair) LOF -> breast cancer, ovarian cancer, prostate cancer
why can sequencing cancer genomes be useful
identify cancer driver genes, analyse types of mutations present, possibly reveal environmental factors (e.g. UV light in skin cancer)
can show different genetics of different evolutions of the tissue (e.g. benign tumour vs cancer vs normal tissue)
-can show the genetic changes needed for metastasis (seq the primary tumour and metastasis)
give an example of driver genes associated w a specific and all cancer types
APC mutations associated w colon cancers but not others, RAS & TERT associated w many cancer types (only a few genes are associated w MANY cancer types)
describe the development of melanomas
normal monocytes -> mutated BRAF (proto oncogenes) ->benign naevus -> TERT mutated (proto oncogenes) -> dysplastic naevus -> loss of CDN2A &/ or TP53 -> melanoma
describe organotropism
most cancers non-random site for metastasis (e.g. prostate cancer -> bone meta)
what did the trial of >25,000 patients with advanced cancer
found copy no. changes in some some genes between the primary & secondary tumour (KRAS, MYC), while tp53 most commonly mutated in secondary tumour
no single mutation / group predicted where it would metastasise
describe the stages of metastasis
after mutations -> infiltration -> intravasation -> circulation -> extravasation -> micrometastasis -> metastatic colonisation
give examples of how epigenetic cause cancer
epigenetic changes have been found between the primary and secondary tumour in pancreatic cancer (pancreatic intraepithelial neoplasia)
breast cancer: methylates E-cadherin promoter (increases metastasis),
demethylates SAAs promoter- binds NF-KB & C/EBP (increases metastasis),
modified mRNA binds e.g. promoter & recruits e.g. LSD complex (increases metastasis),
demethylation of H3 causes stopping of TGF B1 transcription (decreases metastasis)
what are some cancer treatments and how do they work
chronic myeloid leukaemia- caused by BCR-ABL kinase fusion -> so tyrosine kinase inhibitor developed (imatinib methylate targets BCR-ABL1 oncoprotein)
allogenic stem cell transplantation & interferon alpha treatment -> improved clinical outcomes
PD-1 /PDL-1 inhibition (Nivolumab inhibit PD1 for melanoma) developed -> so receptor on tumour cell & T cell blocked & T cells kills tumour
describe HER2 and its role in breast cancer and treatment
HER2= tyrosine kinase receptor (has no ligand, activates signalling by forming a heterodimer w other growth factor receptors- unlike HER, these do have ligands) ), amplified in breast/gastric cancer -> increased cell signalling & proliferation
herceptin inhibits HER2 -but only works in pts w amplified HER2 (so tested b4 administered)
describe petos paradox
greater number of cells & longer lifespans but don’t have higher cancer incidence
why do mole rats have fewer mutations from similarly sized animals & has life span & similar mutation rates to giraffes
high hyaluronan - protects from ROS damage, prevent hyperplasia & have lncRNAs strongly co-expresed with potential tumour suppressors
when irradiated: more rapid DNA damage/ repair response (NMRs enhancers mechanism)- so its proposed that high activity of SIRT6 resorting in modified PARP & more efficient DNA repair
what challenged petos paradox
a study that looked at amphibians, birds, mammals & reptiles
larger animals generally did develop more neoplasia/ malignancy
however the rate of evolutionary body mass increase (path wise rate) was associated w less neoplasia malignancy- so body size evolution in higher rates may reflect more superior anti-cancer mechanisms
why is it though elephants less likely to get cancer
high no. o53 pseudogenes= hypersensitive to stress (apoptosis not senescence)
high no. LIF pseudogenes- pro apoptotic
accelerated regions at VRK2-FANCL-BCL11A locus
increased p21 expression & cell deaty 2x more sensitive than human cells in response to ironing radiation & doxorubicin
why is it thought whaLes are resistant to cancer
have 20x of TP53 & increase copy no. of genes in p53 path such as TSC2 & TP73