Food Analysis Final
1/45
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
46 Terms
Define: Chromatography
A technique for separating components of mixtures as they are carried by a mobile phase through a stationary phase. Compounds are separated based on differences in how strongly they interact with each phase.
Define: Mobile phase
The moving material (liquid or gas) that carries the sample through the stationary phase. In HPLC it is a liquid; in GC it is a gas (e.g., He, N2, H2).
Define: Stationary phase
The phase that does not move — it may be a solid support (e.g., silica) or a liquid coated/bonded onto a solid support. Compounds interact with it and are slowed down based on their affinity.
Define: Adsorbent
The stationary phase used in adsorption chromatography — a solid that compounds stick to on its surface. Common adsorbents: silica gel (-Si-OH) and alumina (-Al-OH).
Define: Eluant
The mobile phase after it has passed through the column and exited (eluted) from it. Also called the eluate.
The Chromatography Triangle — Three Major Components
Separation depends on the interaction of THREE components working together:
• The compounds to be separated — their polarity, molecular size, charge, and volatility determine how they interact with the other two components.
• The stationary phase — its polarity and chemistry determine which compounds are retained and for how long. Compounds with higher affinity for the stationary phase move more slowly.
• The mobile phase — its polarity and eluting strength determine how well it competes with the stationary phase. Compounds with higher affinity for the mobile phase move faster. Changing any one side of the triangle changes the entire separation. All three must be optimized together
Adsorption Chromatography (Liquid/Solid):
The stationary phase is a solid adsorbent (e.g., silica gel with -Si-OH groups, alumina with -Al-OH).
Compounds are separated by how strongly they adsorb onto the solid surface via hydrogen bonding, dipole-dipole, and van der Waals forces.
More polar compounds adsorb more strongly on polar adsorbents and elute later.
Normal-phase TLC and column chromatography are the main examples.
Partition Chromatography (Liquid/Liquid):
A liquid is immobilized on a solid matrix (e.g., silica gel) to form a liquid stationary phase.
Compounds separate based on their relative solubility (partitioning) between the mobile phase liquid and the stationary phase liquid — similar to liquid-liquid extraction.
Gas-liquid chromatography (GC) is the primary example: compounds partition between the carrier gas and the liquid stationary phase coated on the column.
Ion Exchange Chromatography and their 3 applications :
Ions in the liquid mobile phase reversibly exchange with ions on an ionic solid support. Compounds are separated by charge and binding affinity to the ionic resin.
Applications:
(1) separating ionic from nonionic compounds,
(2) separating cations from anions,
(3) separating mixtures of similarly charged ions by their relative binding strength
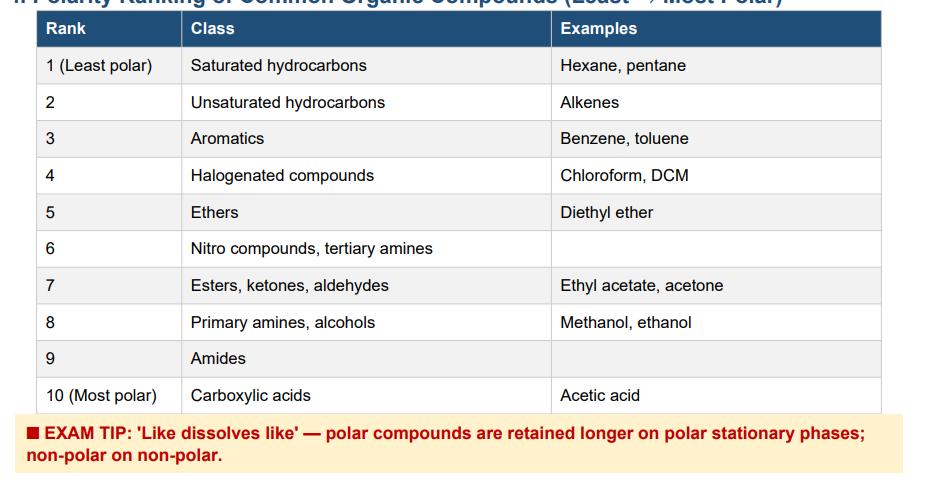
Rank common organic compounds in order of polarity (least to most polar).
Saturated hydrocarbons (hexane) → Unsaturated hydrocarbons (alkenes) → Aromatics (benzene) → Halogenated compounds (chloroform) → Ethers → Nitro compounds / tertiary amines → Esters, ketones, aldehydes (ethyl acetate, acetone) → Primary amines, alcohols (methanol) → Amides → Carboxylic acids (most polar)
Adsorption vs. Partition Chromatography (Side-by-Side)
Adsorption: Stationary phase is a SOLID (silica, alumina). Compounds interact directly with the solid surface. More polar compounds stick more to polar adsorbents. Used in normal-phase TLC and column chromatography.
Partition: Stationary phase is a LIQUID immobilized on a solid. Compounds dissolve into the liquid stationary phase and partition based on relative solubility. Used in GC (gas-liquid chromatography).
Key difference: Adsorption = solid stationary phase, compounds adsorb to surface. Partition = liquid stationary phase, compounds dissolve into it
6. Rf Value in TLC
Rf = distance the solute moves ÷ distance the solvent front moves
Rf ranges from 0 to 1. It is constant for a given compound under the same solute/sorbent/solvent conditions. Used to identify compounds by comparing with known standards run on the same TLC plate.
• Normal-phase TLC (silica): Non-polar compounds have HIGHER Rf (move farther). Polar compounds have LOWER Rf (retained more strongly by polar silica).
• Reversed-phase TLC (C18): Polar compounds have HIGHER Rf. Non-polar compounds have LOWER Rf.
• Detection: UV light (fluorescent indicator) or iodine vapor.
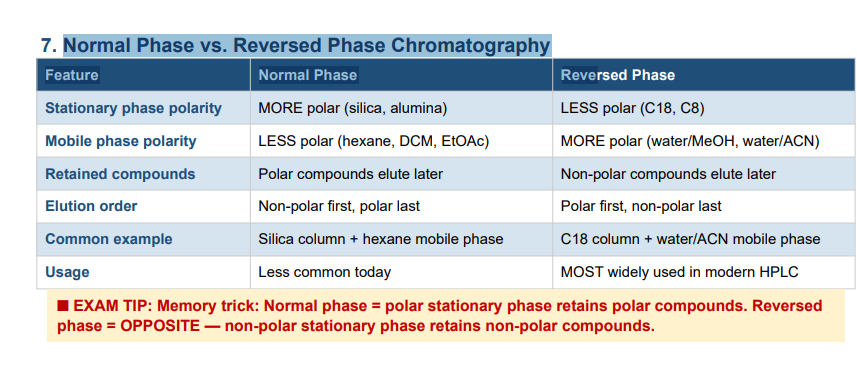
Normal Phase vs. Reversed Phase Chromatography Feature Normal Phase Reve
Normal phase: Stationary phase is MORE polar (e.g., silica, alumina); mobile phase is LESS polar (organic solvents). Polar compounds are retained longer; non-polar compounds elute first.
Reversed phase: Stationary phase is LESS polar (e.g., C18, C8); mobile phase is MORE polar (e.g., water/acetonitrile). Non-polar compounds are retained longer; polar compounds elute first. Reversed phase is the most widely used mode in modern HPLC.
Basic HPLC Instrumentation (in order)
Solvent reservoir (mobile phase)
Pump — delivers mobile phase at constant flow rate and high pressure
Controller — controls pump, gradient program, oven temperature • Injector (sample loop) — introduces a measured volume of sample
Column — where separation occurs
Detector — UV, fluorescence, RI, ECD, MS
Recorder / Data system — records the chromatogram
HPLC Column Polarity Ranking (Most → Least Polar)
Si > NH2 > CN > C1 > C8 > C18 > C30
Silica (Si) is most polar — bare -Si-OH groups.
Longer carbon chains (C18, C30) are most non-polar. Used in reversed-phase mode with polar mobile phases (water/MeOH or water/ACN
3. Guard Column
What it is: A short column packed with the same stationary phase as the analytical column, placed just before it.
Why use it: Protects the expensive analytical column from particulates, strongly retained compounds, and contaminants. When fouled, it is simply replaced cheaply instead of replacing the analytical column.
Isocratic vs. Gradient Elution
Isocratic: Mobile phase composition is CONSTANT throughout the run. Best for samples where all compounds have similar polarities and retention times.
Gradient: Mobile phase composition CHANGES over time (e.g., increasing % organic solvent). Used when the sample contains compounds with a wide range of polarities — gradient can separate both polar and non-polar compounds in a single run
Special Concerns for Sample Preparation Before HPLC
• Sample must be a clear, particle-free solution — filter through 0.45 µm membrane for 5 µm column; 0.2 µm for 3 µm column
• Sample must be soluble in the mobile phase
• The sample solvent must be miscible with the mobile phase
• The polarity of the sample solvent should equal or be weaker than the mobile phase (to avoid peak distortion)
HPLC Detectors
1. UV-Visible absorption detector
2. Fluorescence detector
3. Refractive Index (RI) detector
Electrochemical Detector (ECD)
Mass Spectrometry (MS) detector
1. UV-Visible absorption detector: Measures absorption of UV-Vis light (190–800 nm) as analyte passes through a flow cell. Follows Beer's Law. Sensitive to aromatics and conjugated pi-bond systems. Most common HPLC detector.
2. Fluorescence detector: Excites compounds with UV light; measures longer-wavelength fluorescence emitted. Higher sensitivity and selectivity than UV. Best for compounds with conjugated cyclic structures (PAHs, vitamins, aflatoxins).
3.Refractive Index (RI) detector: univeral detector; measures difference in refractive index between eluent and pure mobile phase. Used for sugars (no UV absorption). Less sensitive than UV.
4. Electrochemical Detector (ECD) : Measures current from oxidation/reduction of compounds. Used for 8-OH-dG, catecholamines, vitamin C.
5. Mass Spectrometry (MS) detector: Most informative; identifies unknowns by mass spectrum. Used for LC-MS.
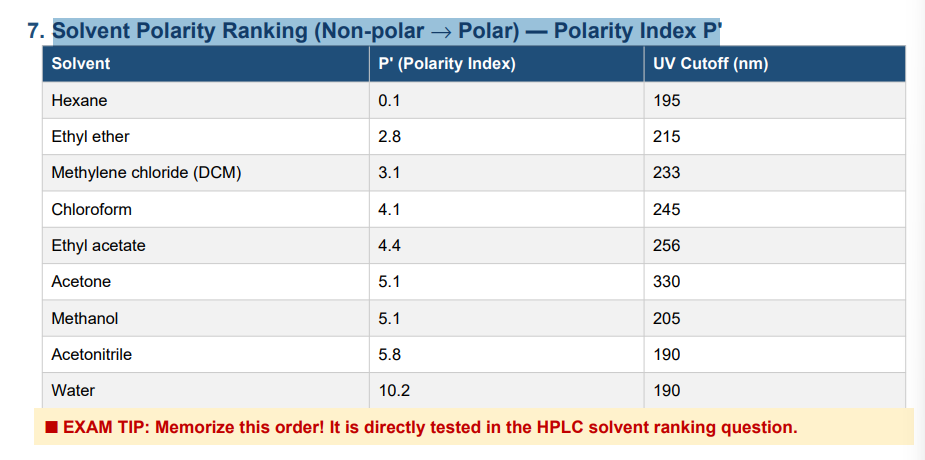
Solvent Polarity Ranking (Non-polar → Polar) — Polarity Index P
UV Cutoff
acetonitrile vs Ethyl acetate vs chloroform vs Methanol
The UV cutoff is the wavelength at which a solvent's absorbance equals 1 AU (10% transmittance) in a 1 cm path cell using water as reference. You cannot detect analytes below the solvent's UV cutoff — the solvent absorbs the light instead.
• Acetonitrile: 190 nm — excellent, very low cutoff
• Methanol: 205 nm — very good
• Chloroform: 245 nm — cannot use UV detection below 245 nm
• Ethyl acetate: 256 nm
Silica Column Mobile Phase Restriction
Water CANNOT be used with a silica (Si) column. Water irreversibly damages the silica stationary phase by stripping the -Si-OH surface. Silica columns are for normal-phase mode only — use organic solvents (hexane, DCM, ethyl acetate, etc.).
■ EXAM TIP: Silica = normal phase = organic solvents ONLY in mobile phase. No water!
What type of chromatography is gas-liquid chromatography (GLC) based on?
Gas-liquid chromatography is based on partition chromatography.
The stationary phase is a non-volatile liquid coated on or bonded to the column walls.
Compounds partition between the gas mobile phase and the liquid stationary phase based on their relative volatility and solubility in the liquid phase.
Major Components of a GC System
• Gas supply — carrier gas (He, N2, or H2); for GC-FID also hydrogen and air and make-up gas
• Injector — introduces and vaporizes the sample (inject 1–3 µL)
• Column (packed or capillary) inside the column oven
• Column oven — controls temperature and temperature programming
• Detector — FID, TCD, or MS
• Recorder / Data system
What Type of Chromatography is GLC Based On?
Gas-liquid chromatography is based on partition chromatography.
The stationary phase is a non-volatile liquid coated on or bonded to the column walls.
Compounds partition between the gas mobile phase and the liquid stationary phase based on their relative volatility and solubility.
Principle of Separation in Gas-Liquid Chromatography
The sample is vaporized and swept by an inert carrier gas through the column. Compounds distribute (partition) between the gas phase and the liquid stationary phase:
• Higher affinity for stationary phase (more soluble, less volatile) → retained longer → elute later
• Lower affinity for stationary phase (more volatile, less soluble) → travel faster → elute earlier
Temperature also affects separation — higher oven temperature increases volatility and decreases retention time
Gases Needed for GC-FID with Capillary Column (Fatty Acid Analysis in Butter)
• Carrier gas (He or N2) — moves sample through column
• Hydrogen (H2) — fuel gas for the FID flame
• Air — oxidant/support gas for the FID flame
• Make-up gas (He or N2) — additional inert gas at detector to maintain optimal flow
Major Concerns in Column Selection for GC
• Polarity — 'like dissolves like'; match column polarity to analyte polarity
• Efficiency — total number of theoretical plates; longer = better resolution
• Stationary phase content and film thickness — thicker film = more retention but more bleed
• Temperature stability — column must be stable at required analysis temperatures
Which Column Is More Polar? Poly(dimethylsiloxane) or Poly(50% diphenyl/50% dimethylsiloxane)?
Poly(50% diphenyl/50% dimethylsiloxane) is MORE POLAR than poly(dimethylsiloxane).
Diphenyl groups are more polar than methyl groups — more phenyl substitution = more polar column.
Polarity order (least → most polar): Poly(dimethylsiloxane) < Poly(5% diphenyl) < Poly(20% diphenyl) < Poly(35% diphenyl) < Poly(50% diphenyl) < Poly(alkylene glycol) < Poly(ethylene glycol/Carbowax 20M)
Principle of FID (Flame Ionization Detector)
Compounds eluting from the column are burned in a hydrogen/air flame.
Carbon-containing compounds (C-C and C-H bonds) produce ions and electrons during combustion, generating an electrical current between a collector electrode and a jet tip.
This current is measured and is proportional to the number of carbon atoms.
Universal detector for organic carbon-containing compounds
Sensitivity: 10–100 pg
Does NOT respond to H2O, CO2, NO2, or H2S
Gas supply required: carrier gas + hydrogen + air + make-up gas
Why Must Fatty Acids and Sugars Be Derivatized Before GC?
Fatty acids and sugars are too polar and non-volatile in native form — their -COOH and -OH groups form hydrogen bonds, making them non-volatile and thermally unstable at GC temperatures.
Fatty acids → methylated to fatty acid methyl esters (FAMEs) by acid-catalyzed esterification with methanol. FAMEs are volatile and non-polar — compatible with GC.
Sugars → converted to trimethylsilyl (TMS) derivatives using BSA or BSTFA reagents, replacing -OH groups with TMS groups to reduce polarity and increase volatility.
How to Identify Compounds in a GC Chromatogram
• Retention time — compare with a known standard run under identical conditions
• Relative retention time — ratio of adjusted retention times relative to a reference compound
• Spiking — add a known standard to the sample; the target peak increases if the compound is present
• GC-MS — mass spectrometer as detector; computer searches spectral library for structural match
Differences Between Compounds Analyzed by GC vs. HPLC
GC:
Compounds must be volatile and thermally stable.
Best for non-polar to moderately polar small molecules (fatty acid esters, flavor volatiles, pesticides, hydrocarbons, alcohols).
Cannot analyze ionic, thermally labile, or very high molecular weight compounds.
HPLC:
Compounds do NOT need to be volatile — only need to be soluble in the mobile phase.
Can analyze non-volatile, thermally labile, ionic, polar, and high molecular weight compounds (proteins, carbohydrates, vitamins, phenolics, amino acids, nucleotides).
External Standard vs. Internal Standard
External Standard:
A separate solution of known concentration is run independently.
A calibration curve (peak area vs. concentration) is built.
Simple but does NOT correct for injection volume variability or sample losses during preparation.
Internal Standard:
A known amount of a compound NOT present in the sample is added directly to the sample BEFORE extraction/preparation.
Quantification uses the ratio of analyte peak area to internal standard peak area.
Corrects for losses during extraction, injection variability, and instrument drift — much more accurate.
Advantages and Applications of Internal Standard in GC/HPLC and requirements of good internal standard
• Corrects for variable injection volumes
• Corrects for analyte losses during sample extraction and preparation
• Corrects for instrument drift or signal instability over time
• Improves precision and accuracy in complex food matrices
Requirements for a good internal standard: chemically similar to analyte; elutes near analyte but does not co-elute; absent from the real sample; stable under analysis conditions
Explain the principle of the Cellular Antioxidant Activity (CAA) assay.
The CAA assay measures antioxidant activity inside living HepG2 human liver cells. Steps: (1) Seed HepG2 cells; incubate 24 hr. (2) Treat cells with DCFH-DA (a fluorescent probe precursor) + antioxidant compounds for 1 hr — DCFH-DA enters cells and is hydrolyzed by cellular esterases to DCFH (non-fluorescent, trapped inside). (3) Add ABAP (peroxyl radical generator) for 1 hr. (4) ABAP-generated peroxyl radicals oxidize DCFH → DCF (fluorescent). Antioxidants inside the cell prevent this oxidation, reducing DCF fluorescence. (5) Measure fluorescence at Ex 485 nm / Em 538 nm. CAA unit = 1 – (Sample Area / Control Area). Higher value = stronger antioxidant.
Describe the advantages of CAA assay over traditional chemical antioxidant activity assays.
Traditional test-tube assays (ORAC, DPPH, FRAP, TEAC) do NOT account for:
• Bioavailability and cellular uptake — a compound that works in a test tube may not enter cells
• Cellular metabolism — compounds may be converted to more or less active forms inside cells
• Partitioning and location within cellular compartments
• Physiological conditions (cellular enzymes, pH, other biomolecules)
• Tissue/cellular concentrations actually achieved in vivo
The CAA assay uses living cells, making it far more biologically relevant for predicting in vivo antioxidant activity.
Describe the principle of Atomic Emission Spectroscopy (AES / ICP-OES).
The sample is introduced into an inductively coupled plasma (ICP) — an extremely hot argon plasma (~7,000–10,000°C) that thermally excites atoms to high energy states. When excited atoms return to ground state, they emit light at characteristic wavelengths specific to each element. The emitted light is detected by a spectrometer. ICP-OES can measure multiple elements simultaneously, making it faster and more comprehensive than AAS. Used for mineral analysis in ash samples.
Define: Antibody
A protein (immunoglobulin) produced by the immune system that specifically recognizes and binds to a particular antigen (target molecule) with high specificity and affinity. In immunoassays, antibodies are the key detection molecules — they bind the compound of interest to generate a measurable signal.
A molecule (typically a protein, polysaccharide, or other macromolecule) that is specifically recognized and bound by an antibody.
Besides nutritional quality, give two examples of why it is important to measure mineral content of foods.
(1) Food safety — toxic metal contamination: Lead (Pb) or cadmium (Cd) in leafy greens or grains can indicate soil or water contamination from industrial sources. Monitoring ensures levels are below regulatory limits to protect consumers.
(2) Food processing environment quality: Monitoring iron (Fe) or copper (Cu) levels in water or equipment contact surfaces prevents catalytic oxidation of lipids and ensures equipment is not corroding into food products.
Describe the advantages and application of an internal standard in GC or HPLC analyses.
Advantages: (1) Corrects for variable injection volumes. (2) Corrects for analyte losses during sample extraction and preparation. (3) Corrects for instrument drift or signal instability. (4) Improves precision and accuracy over external standards in complex food matrices.
Requirements for a good internal standard: Chemically similar to the analyte; elutes near the analyte but does not co-elute; absent from the real sample; stable under analysis conditions.
Define: Mass spectrum
Draw/list the basic instrumentation for a Mass Spectrometry (MS) system.
Sample Introduction (direct injection, direct probe, or GC/HPLC interface) → Ion Source / Ionization (EI or CI — ionizes and fragments the molecule) → Mass Analyzer (separates ions by m/z: quadrupole or ion trap) → Detector (measures separated ions) → Data System (records spectrum, searches library)