Metabolic Final Exam Prep
1/38
Earn XP
Description and Tags
40 vocabulary-style flashcards covering neonatal metabolic disorders, enzyme deficiencies, screening protocols, and genetic inheritance patterns mentioned in the lecture transcript.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
39 Terms
A 5-day-old female is admitted to the neonatal intensive care unit for poor feeding, weakness, lethargy and respiratory distress. Labs are drawn just before IV fluids are started, including complete blood count, electrolytes, lactate, ammonia, urine organic acids, plasma amino acids, acylcarnitine profile and plasma total and free carnitine. The baby is made NPO (nothing by mouth), is intubated and started on mechanical ventilation due to the respiratory distress. The complete blood count, electrolytes, glucose, lactate and ammonia are normal. The other labs are pending. Chest X-ray is performed, which shows a massively enlarged heart. This baby is MOST likely to respond to the following type of treatment:
Enzyme replacement therapy
High-concentration dextrose and fluids
Protein-restricted diet
Enzyme replacement therapy
A 1-day-old female is critically ill due to a complex congenital heart defect and requires a blood transfusion. Newborn screening is collected the following day. Newborn screening results will be LEAST reliable for the following disorder:
Galactosemia
Methylmalonic acidemia
Phenylketonuria
Tyrosinemia
Galactosemia
2-year-old female is referred to Metabolic clinic from Endocrinology for a history of hypoglycemia for the past 6 months. The family has been monitoring her blood glucose levels carefully at home. She consistently becomes hypoglycemic after 5-6 hours of fasting. Some basic labs already done by Endocrinology were remarkable for ketosis and elevated creatine kinase. An echocardiogram was done that showed mild cardiomyopathy. You are suspicious for a glycogen storage disorder. You recommend some additional labs including electrolytes, lactate, uric acid, and lipid panel. You are MOST likely to detect an elevated blood lactate level at the following time point:
1 hour after a meal
4 hours after a meal
8 hours after a meal
12 hours after a meal
1 hour after a meal
A 35-year-old female presents to Genetics clinic, brought by her parents, for a history of severe intellectual disability. She was born in a small town outside of the United States. Pedigree is notable for consanguinity. Her parents are first cousins. You notice that the patient is microcephalic, has a skin rash, has much lighter skin and hair pigmentation than her parents, and is emanating a strong mousy odor. The following amino acid is MOST likely to be deficient:
Alanine
Phenylalanine
Tyrosine
Tryptophan
Tyrosine
A 35-year-old female presents to Genetics clinic, brought by her parents, for a history of severe intellectual disability. She was born in a small town outside of the United States. Pedigree is notable for consanguinity. Her parents are first cousins. You notice that the patient is microcephalic, has a skin rash, has much lighter skin and hair pigmentation than her parents, and is emanating a strong mousy odor.
You are MOST likely to recommend the following treatment:
Branched chain amino acid supplementation
Carnitine supplementation
Large neutral amino acid supplementation
MCT oil supplementation
Large neutral amino acid supplementation
Which of the following BEST explains the mechanism for tachypnea in a neonate with Citrullinemia, experiencing metabolic decompensation?
Ammonia causes metabolic alkalosis
Ammonia causes respiratory alkalosis
Orotic acid causes metabolic acidosis
Orotic acid causes respiratory acidosis
Ammonia causes respiratory alkalosis
You are called by the California Newborn Screening Program with four abnormal newborn screening results. The first is positive for maple syrup urine disease in a 5-day-old male, born at 29 weeks gestation, admitted to the NICU and receiving total parenteral nutrition (TPN). The second is positive for phenylketonuria in a 4-day-old male, born at 40 weeks gestation, discharged home on day-of-life 2. The third is positive for short-chain acyl co-A dehydrogenase deficiency in a 3-day-old female, born at 35 weeks gestation, admitted to the observation nursery to feed and grow. The fourth is positive for very long chain acyl-CoA dehydrogenase in a 4-day-old female, born at 37 weeks gestation, discharged home on day-of-life 2. It is MOST important that you get in IMMEDIATE contact with the family of the baby with newborn screening results positive for the following disorder:
Maple syrup urine disease
Phenylketonuria
Short chain acyl-CoA dehydrogenase deficiency
Very long chain acyl-CoA dehydrogenase deficiency
Very long chain acyl-CoA dehydrogenase deficiency
You see a 2-week old male in clinic for a history of positive newborn screen. On family history, the patient has a full sister with disproportionately long arms and legs and mild intellectual disability. A 15-year-old first cousin (son of maternal uncle) has a history of intellectual disability and recently had a stroke. Parents do not report consanguinity but have the same surname and are from the same small town (population 1,000).
Your patient’s newborn screen is MOST likely positive for the following disorder:
Alkaptonuria
Cystinuria
Homocystinuria
Hyperoxaluria
Homocystinuria
You see a 2-week old male in clinic for a history of positive newborn screen. On family history, the patient has a full sister with disproportionately long arms and legs and mild intellectual disability. A 15-year-old first cousin (son of maternal uncle) has a history of intellectual disability and recently had a stroke. Parents do not report consanguinity but have the same surname and are from the same small town (population 1,000). You are MOST likely to recommend a therapeutic trial with the following vitamin:
Vitamin B1 (Thiamine)
Vitamin B2 (Riboflavin)
Vitamin B6 (Pyridoxine)
Vitamin B7 (Biotin)
Vitamin B6 (Pyridoxine)
In cases of organic acidemias, the MOST likely primary abnormality leading to rapid breathing is:
Metabolic acidosis
Metabolic alkalosis
Respiratory acidosis
Respiratory alkalosis
Metabolic acidosis
The term “Leigh Disease” implies knowledge of a specific genetic defect. True or False
False
Defective glycogenolysis AND gluconeogenesis is associated with the following glycogen storage disorder:
I
III
IV
V
I
A 20-year-old male is referred to Genetics clinic by Pulmonology for a history of lung disease, bone pain and hepatosplenomegaly. He is also followed by Hematology for a history of anemia and thrombocytopenia. Bone marrow biopsy was done recently and was remarkable for abnormal macrophages, which had increased lipid content. X-rays are MOST likely to show the following:
Biconvex vertebrae
Cardiomegaly
Focal lytic lesions
Punctate calcifications
Focal lytic lesions
A 4-month-old female is seen in the ER at 7am for decreased activity level, sweating and irritability. She last fed a full bottle of formula at 2am. Prior to this episode, she had done generally well since birth, feeding well and with a normal number of wet diapers. She just started sleeping for longer intervals. While she is in the waiting room, she becomes unresponsive. Rapid glucose check shows a blood glucose level of “0”, or below the limit of detection. Which of the following types of disorders is MOST likely?
Fatty acid oxidation disorder
Glycogen storage disorder
Glycogen storage disorder
A 4-month-old female is seen in the ER at 7am for decreased activity level, sweating and irritability. She has severe nasal congestion (stuffiness), rhinorrhea (runny nose), and has fed poorly for the past two days, crying with every feed and taking minimal volume. While she is in the waiting room, she becomes unresponsive. Rapid glucose check shows a blood glucose level of “0”, or below the limit of detection. Which of the following types of disorders is MOST likely?
Fatty acid oxidation disorder
Glycogen storage disorder
Fatty acid oxidation disorder
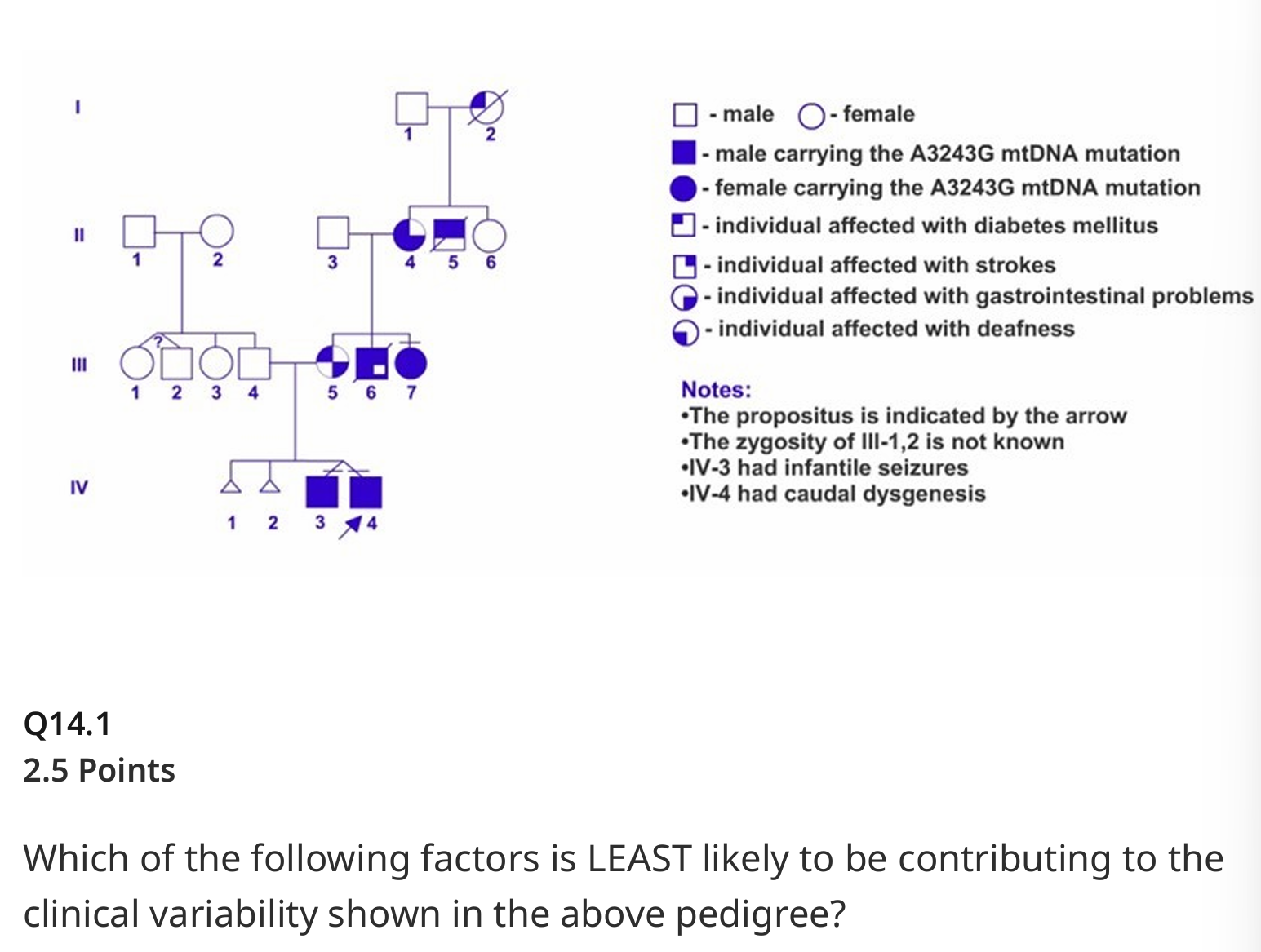
Which of the following factors is LEAST likely to be contributing to the clinical variability shown in the above pedigree?
Environmental factors
Heteroplasmy
Skewed X-inactivation
Threshold effect
Skewed X-inactivation
A female neonate is noticed upon delivery to have rhizomelic limb shortening, small rib cage and cataracts. Within hours after birth, she requires intubation and mechanical ventilation due to respiratory failure. Once stabilized, skeletal survey is done which shows punctate calcifications in cartilage with epiphyseal and metaphyseal abnormalities, and coronal clefts of the vertebral bodies. She is MOST likely to have a disorder of the following type:
Lysosome biogenesis disorder
Lysosome single enzyme defect
Peroxisome biogenesis disorder
Peroxisome single enzyme defect
Peroxisome biogenesis disorder
A 6-year-old male is seen in Metabolic clinic for a history of galactosemia. He was referred by an Emergency Department physician after been seen for an unrelated head injury. Upon history, his mother told the ED physician, “He has galactosemia. Someone told me this when he was born.” The patient does not have any significant past medical history aside from the recent head injury. He is in first grade, regular classes, doing well. He has never been on a special diet and in fact loves milk and cheese. GALT molecular sequence analysis is MOST likely to show the following:
Compound heterozygosity for a pathogenic variant and a Duarte variant
Compound heterozygosity for two pathogenic variants
Homozygosity for a Duarte variant
Homozygosity for a pathogenic variant
Compound heterozygosity for a pathogenic variant and a Duarte variant
Newborn screening results are flagged positive for a 5-day-old female, showing near-absent activity of galactose-1-phosphate uridyltransferase. She is currently admitted to the neonatal intensive care unit due to hyperbilirubinemia and is receiving phototherapy. She is otherwise well, breathing comfortably, breastfeeding well, with good urine output and normal stool.
What is the BEST next step?
Obtain red blood cell gal-1-p level
Obtain urine reducing substances
Switch to a fructose-free formula
Switch to a lactose-free formula
Switch to a lactose-free formula
Newborn screening results are flagged positive for a 5-day-old female, showing near-absent activity of galactose-1-phosphate uridyltransferase. She is currently admitted to the neonatal intensive care unit due to hyperbilirubinemia and is receiving phototherapy. She is otherwise well, breathing comfortably, breastfeeding well, with good urine output and normal stool.
Which clinical outcome is LEAST likely to be affected by dietary treatment?
Cataracts
Sepsis
Liver failure
Ovarian failure
Ovarian failure
A 10-year-old male, Tommy Smith, presents to Genetics clinic for a history of molecularly-confirmed X-linked adrenoleukodystrophy. He was followed previously by Metabolic at Johns Hopkins, and the family just moved to California two days ago. The patient has two full younger brothers, Danny, age 7-years and Billy, age 5-years. Upon further questioning, his parents state that both younger brothers underwent targeted mutation analysis, and both were found to be positive for the familial mutation. You do not have the original reports for review, but the parents state, “we have tons of records at home. All of our boys had brain MRI’s and neuropsychological testing two weeks ago.” Physical examination of Tommy shows decreased understanding of questions and poor coordination. Physical examinations of Danny and Billy are entirely normal. You request outside records, which the parents bring the following day. Genetic testing results confirm presence of a known pathogenic mutation in all three children. Further review shows that Tommy has severe intellectual disability and brain MRI with white matter abnormalities. Danny has a normal IQ and brain MRI with white matter abnormalities. Billy has a normal IQ and normal brain MRI. You are MOST likely to recommend immediate hematopoetic stem cell transplantation evaluation for the following individual(s):
Billy
Danny
Tommy
Billy and Danny
Danny and Tommy
Billy, Danny and Tommy
Danny
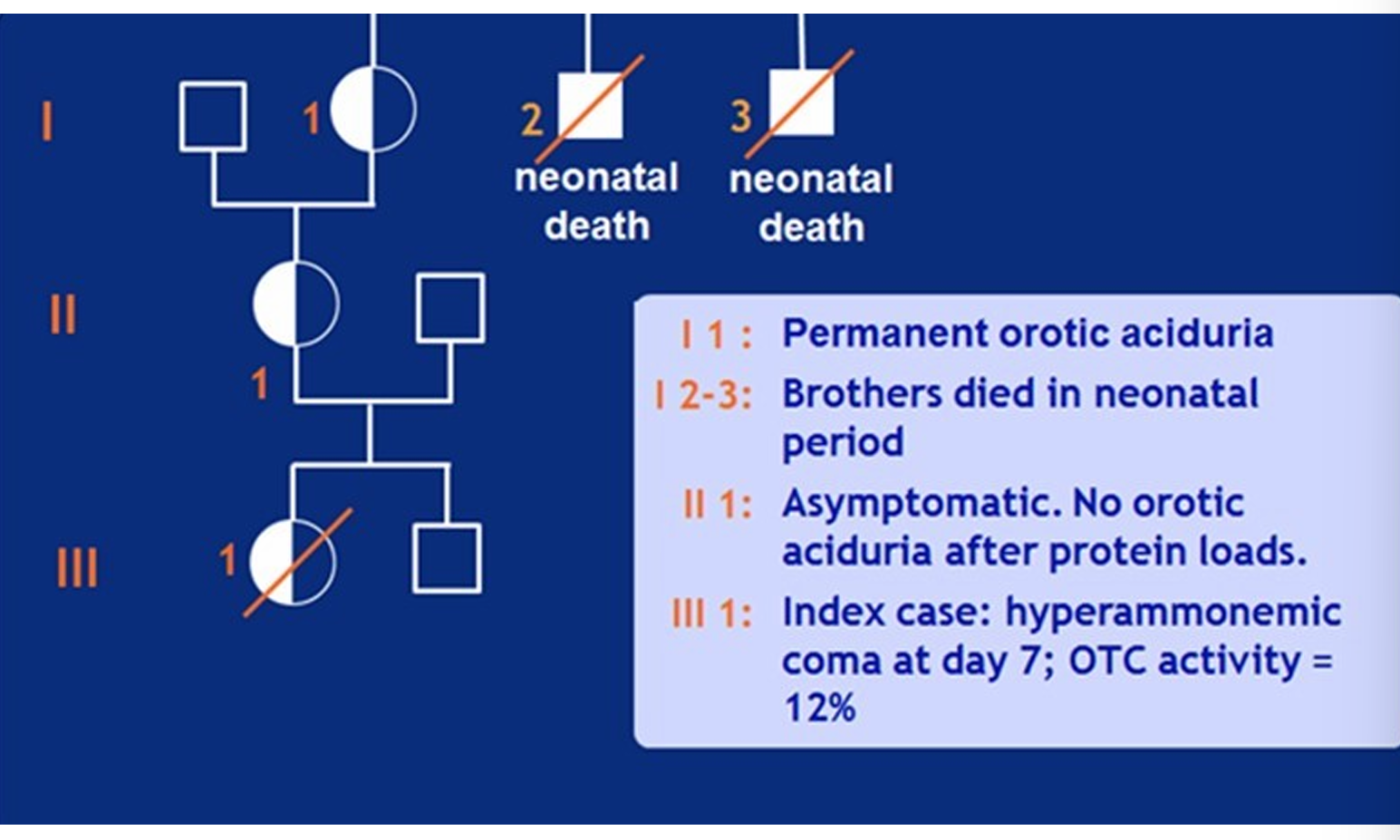
Which of the following factors is MOST likely to be contributing to the clinical variability shown in the above pedigree?
Germline mosaicism
Heteroplasmy
Skewed X-inactivation
Threshold effect
Skewed X-inactivation
A 3-year-old male is seen in Metabolic clinic for a history of profound hypoglycemia when fasting longer than 4 hours. His cheeks appear chubby and tight. He has an enlarged liver and spleen. You are suspicious for glycogen storage disease type 1A versus glycogen storage disease type 1B. Presence of which of the following lab abnormalities is MOST likely to help you distinguish between these two disorders?
Decreased urinary uric acid
Elevated blood lactate
Neutropenia
Thrombocytopenia
Neutropenia
A 35-year-old female presents to Metabolic clinic, with a known history of phenylketonuria due to phenylalanine hydroxylase deficiency. Her care has lapsed since age 22 years when she lost CCS insurance. She just recently regained insurance, a Medi-Cal HMO plan, and established care with a primary care physician. Today she complains of anxiety, depression, confusion and fatigue. She is not following any dietary restrictions. Her primary care physician sent a plasma phenylalanine level 2 weeks ago, which was 1800 µmol/L. At the end of the visit, she discloses that she is 5 weeks pregnant and desires to continue the pregnancy. She cannot afford to pay for low-protein medical foods while awaiting insurance approval.
You are MOST likely to recommend the following next steps in management:
Initiate dietary treatment inpatient, check phenylalanine three times per week
Initiate dietary treatment outpatient, check phenylalanine once per month
Initiate dietary treatment outpatient, check phenylalanine once per week
Discharge patient from Metabolic care, since dietary treatment is no longer necessary
Initiate dietary treatment inpatient, check phenylalanine three times per week
A 35-year-old female presents to Metabolic clinic, with a known history of phenylketonuria due to phenylalanine hydroxylase deficiency. Her care has lapsed since age 22 years when she lost CCS insurance. She just recently regained insurance, a Medi-Cal HMO plan, and established care with a primary care physician. Today she complains of anxiety, depression, confusion and fatigue. She is not following any dietary restrictions. Her primary care physician sent a plasma phenylalanine level 2 weeks ago, which was 1800 µmol/L. At the end of the visit, she discloses that she is 5 weeks pregnant and desires to continue the pregnancy. She cannot afford to pay for low-protein medical foods while awaiting insurance approval.
You are LEAST likely to recommend the following treatment:
Large neutral amino acids
Low-protein medical foods
Tetrahydrobiopterin
Tyrosine supplementation
Large neutral amino acids
A 5-year-old male presents to Genetics clinic for developmental delay. Upon further questioning, you note that he reached early milestones like smiling, laughing, cooing, rolling over, on time. He spoke his first words at 12 months and by 2 years had 50 words and spoke in 2-word phrases. Now, at age 5-years, he has fewer words than he did at age 2 years. He has trouble walking long distances and writing. Family history is notable for 3 full siblings, including 2 sisters, age 2 years and 8 years, who are both healthy and developmentally appropriate, and one brother, age 3 years, with mild speech and motor developmental delay. Physical examination of your patient shows frontal bossing, periorbital fullness, broad nasal bridge and tip, and full lips. His parents show you a picture from his 1st birthday, and he looks remarkably different. You obtain a skeletal survey, urine glycosaminoglycans, lysosomal enzyme screen and refer to ophthalmology. The ophthalmologist notes a completely normal exam, with no evidence of corneal clouding.
The disorder present in the patient is MOST likely associated with the following mode of inheritance:
Autosomal dominant
Autosomal recessive
X-linked dominant
X-linked recessive
X-linked recessive
A 5-year-old male presents to Genetics clinic for developmental delay. Upon further questioning, you note that he reached early milestones like smiling, laughing, cooing, rolling over, on time. He spoke his first words at 12 months and by 2 years had 50 words and spoke in 2-word phrases. Now, at age 5-years, he has fewer words than he did at age 2 years. He has trouble walking long distances and writing. Family history is notable for 3 full siblings, including 2 sisters, age 2 years and 8 years, who are both healthy and developmentally appropriate, and one brother, age 3 years, with mild speech and motor developmental delay. Physical examination of your patient shows frontal bossing, periorbital fullness, broad nasal bridge and tip, and full lips. His parents show you a picture from his 1st birthday, and he looks remarkably different. You obtain a skeletal survey, urine glycosaminoglycans, lysosomal enzyme screen and refer to ophthalmology. The ophthalmologist notes a completely normal exam, with no evidence of corneal clouding.
Skeletal survey is LEAST likely to show the following finding:
Bones in the legs that look like tissue paper
Bones in the hands that look like bullets
Ribs that look like oars or paddles
Vertebrae that look like hooks or pears
Bones in the legs that look like tissue paper
Development of the following technology MOST revolutionized newborn screening for inborn errors of metabolism:
Ion mass spectrometry
Isotope mass spectrometry
Tandem mass spectrometry
Thermal mass spectrometry
Tandem mass spectrometry
A 9-month-old female is admitted to the pediatric intensive care unit for new-onset seizures and metabolic acidosis. Plasma amino acids show elevated alanine. Brain MRI with MR spectroscopy is done, showing bilateral, symmetric lesions in the basal ganglia associated with lactate peaks. Sadly, the baby develops respiratory failure, multiorgan failure, and dies three days later. Which of the following could be the cause of the baby’s findings?
Mutations in mitochondrial-encoded mitochondrial proteins
Mutations in nuclear-encoded mitochondrial proteins
Mutations in pyruvate dehydrogenase complex genes
All of the above
All of the above
The neonatal intensive care unit calls you regarding a 1-day-old male with hypotonia, questionable seizure activity and elevated liver enzymes. Physical exam shows dysmorphic facial features including coarse facial features and large anterior fontanelle. Brain MRI is done, showing ventriculomegaly, neuronal migration defect and pachygyria. You suspect a metabolic disorder involving an organelle with the following function:
Breakdown of bile acids
Breakdown of glycosaminoglycans
Synthesis of plasmalogens
Synthesis of very long chain fatty acids
Synthesis of plasmalogens
A 23-year-old female enters military boot camp. The night after the first day of training, she experiences extreme muscle pain in her legs. She feels a little better after a hot shower, takes some Tylenol and manages to fall asleep. The next morning, she does not have the strength to get out of bed by herself. With help she goes to the bathroom and is shocked when she sees that her urine has turned dark brown. Labs show an elevated creatine kinase (CK) of greater than 100,000 units/L (normal 38 176). Which of the following labs is MOST likely to be helpful in this patient’s diagnostic workup?
Plasma acylcarnitines
Plasma amino acids
Urine orotic acid
Urine succinylacetone
Plasma acylcarnitines
A 13-month-old female is admitted to the pediatric intensive care unit for poor feeding and lethargy. She had vomiting and diarrhea for 3 days prior to admission. On the day of admission, the diarrhea improved, but she had minimal wet diapers. The following lab abnormality would raise the MOST suspicion for a disorder of fatty acid oxidation:
Absent urine ketones
Hyperammonemia
Hypoglycemia
Metabolic acidosis
Absent urine ketones
A 5-year-old female presents to Genetics clinic for developmental delay. Upon further questioning, you note that she reached early milestones like smiling, laughing, cooing, rolling over, on time. She spoke her first words at 12 months and by 2 years had 50 words and spoke in 2-word phrases. Now, at age 5-years, she has fewer words than she did at age 2 years. She has trouble walking long distances and writing. Her parents show you a picture from her 1st birthday, and she looks remarkably different. She was admitted to the hospital 6 months prior to this visit for pneumonia. Chest X-ray was done during that admission. It was read by Radiology as consistent with pneumonia, no fractures, and otherwise normal. You look at the images and notice that the ribs look different, with certain sections appearing wider than other sections. While speaking with the family, you notice that something about the patient’s eyes looks “cloudy”. The patient is MOST likely to have a defect in breaking down the following:
Dermatan sulfate
Heparan sulfate
Keratan sulfate
Dermatan sulfate and heparan sulfate
Dermatan sulfate and keratan sulfate
Heparan sulfate and keratan sulfate
Dermatan sulfate and heparan sulfate
A 5-year-old female presents to Genetics clinic for developmental delay. Upon further questioning, you note that she reached early milestones like smiling, laughing, cooing, rolling over, on time. She spoke her first words at 12 months and by 2 years had 50 words and spoke in 2-word phrases. Now, at age 5-years, she has fewer words than she did at age 2 years. She has trouble walking long distances and writing. Her parents show you a picture from her 1st birthday, and she looks remarkably different. She was admitted to the hospital 6 months prior to this visit for pneumonia. Chest X-ray was done during that admission. It was read by Radiology as consistent with pneumonia, no fractures, and otherwise normal. You look at the images and notice that the ribs look different, with certain sections appearing wider than other sections. While speaking with the family, you notice that something about the patient’s eyes looks “cloudy”. The disorder present in the patient is MOST likely associated with the following mode of inheritance:
Autosomal dominant
Autosomal recessive
X-linked dominant
X-linked recessive
Autosomal recessive
You are called by the CA state newborn screening program with results for a 4-day-old female, flagged positive for carnitine transporter defect due to low free carnitine (C0) below the lower cutoff. Confirmatory plasma and urine carnitine, plasma acylcarnitine profile, and urine organic acids are obtained for both the baby and the mother. The baby’s confirmatory testing is essentially normal, however the mother is found to have profoundly low plasma carnitine levels. Her other biochemical laboratory testing is LEAST likely to show findings concerning for the following disorder:
Carnitine palmitoyltransferase I deficiency
Carnitine transporter defect
Medium-chain acyl-CoA dehydrogenase deficiency
Very long-chain acyl-CoA dehydrogenase deficiency
Carnitine palmitoyltransferase I deficiency
A newborn on total parental nutrition (TPN) is MOST likely to have false positive newborn screening results for the following disorder:
Isovaleric acidemia
Maple syrup urine disease
Methylmalonic acidemia
Short chain acyl-CoA dehydrogenase (SCAD) deficiency
Maple syrup urine disease
A 3-year-old female presents to Metabolic clinic to establish care. The family just moved to California from another country. The child’s parents state she screened positive for a metabolic disorder, however they cannot recall the specific disorder, and she is not on any diet or medication treatment. You send some basic metabolic labs that show: normal ammonia, plasma amino acids, and lactate; plasma acylcarnitines with elevated C6, C8 and C10, plasma total and free carnitine both mildly reduced, and urine organic acids with elevated adipic, suberic, sebacic acids, 5-hydroxy hexanoic acid, hexanoylglycine, phenylpropionylglycine, and particularly suberylglycine. You are LEAST likely to recommend the following treatment:
Carnitine supplementation
Cornstarch
Low-fat diet
MCT oil
MCT oil
A 14-month-old female is seen in outpatient Genetics clinic for developmental delay and failure to thrive. Brain MRI done previously shows "white matter changes". She has minimal spontaneous movement and diffuse hypotonia. Physical exam shows epicanthal folds. The disorder you suspect is MOST likely to show this additional finding on physical exam:
Bowed lower extremities
Periumbilical angiokeratomas
Thoracolumbar kyphosis
Wide anterior fontanel
Wide anterior fontanel
A 6-year-old male with a history of intellectual disability presents to the ER with acute neurologic deterioration in the setting of a viral illness. Physical exam shows dystonia. Brain MRI shows decreased signal in bilateral basal ganglia. His mother has a history of early-onset diabetes, headaches and cardiomyopathy of unknown cause. Rapid genome sequencing of the patient and both parents shows a pathogenic variant, 3243A>G in MT-TL1 tRNALeu, in the patient but neither parent. The absence of this pathogenic variant in the patient's mother is MOST likely related to the following:
Environmental effects
Heteroplasmy
Skewed X-inactivation
Threshold effect
Heteroplasmy