Drugs and Behavior - Exam 3
1/125
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
126 Terms
Effects of chronic psychostimulant use on the brain
Reduced cortical/PFC activity
Neuronal cell death/neurotoxicity → e.g. the loss of dopamine neurons
Also reduced neurogenesis (evidenced in animal research)
Behavioral effects & PFC: reduced behavioral inhibition, etc.
PD tolerance/downregulation of D1 & D2 receptors → can rebound to some extent with continued abstinence.
Formation of gaps in the BBB
Effects of chronic psychostimulant: Psychological
Psychotic symptoms ⇒ sensitized effect
Monoamine psychosis: symptoms similar to paranoid schizophrenia.
Delusions: persecutory or grandiose
Hallucinations: sometimes visual (fleeting shadows, flashing colors) or tactile (formication)
Effects of chronic psychostimulant: Physical
Severe physical consequences: Mostly driven by malnutrition and poor hygiene
Significant weight loss/malnutrition: psychostimulants reduce appetite.
Dental issues: bruxism (teeth grinding), dry mouth, tooth wear/decay, periodontal disease → especially due to smoking.
Psychostimulant Dependence & Withdrawal
Crash/comedown anhedonia: period of depression, lethargy ~24 hours after drug cessation.
Likely due to monoamine depletion from extended use.
Compensatory increases in sleeping and eating.
~1 week-several months: anxiety, agitation, mood depression, and drug cravings.
>/=1 year: Long-term cognitive dysfunction —> study of people with methamphetamine use disorder found significant impairments in decision-making w/ concomitant reduction in PFC activity.
Behavioral Approaches: Treatment for Psychostimulant Misuse
CBT: relapse prevention strategies
Contingency management and community reinforcement:
Environmental and lifestyle adjustments + skills training ⇒ replace drug-seeking behavior.
Both of these use operant techniques: reinforce abstinence ⇒ tangible reinforcers (e.g. money, goods, services) or social (e.g. praise)
Pharmacotherapeutic Approaches: Treatment for Psychostimulant Misuse
Currently no good maintenance therapy for psychostimulants
Mirtazapine (increases monoamine levels = reduces withdrawal symptoms) + naltrexone (opioid receptor antagonist = reduces pleasure) ⇒ shown promise as treatment.
Immunization to create antibodies against psychostimulants?
Opioids
Definition: drugs w/ properties similar to opium or its principal psychoactive ingredients (extracts: morphine, codeine, and thebaine)
Strong analgesic (“pain killers”) properties, without anesthetic effects.
Other names: narcotics, narcotic analgesics
Narcoticum (Latin): make stiff/numb
Narkoe (Greek): stupor/sleep
Over the years, the term narcotic has been bastardized by US law enforcement as a name for any illicit drug w/ misuse potential ⇒ developed a pejorative connotation of drug users.
Natural origin of opioids
Poppy Plants “Papaver somniferum.”
Principal natural source of opium → produces only 10 days in its life cycle after petals fall off.
Intentional scratches made on the seedpod exude milky fluid = latex (“opium milk”) → scraped off, compressed into cakes, and dried = opium.
Extracts (psychoactive constituents): Morphine (10%), codeine (2.5%), and thebaine (1%).
Semi-synthetic opioids: Morphine derived
Heroin (diacetyl morphine): Morphine + 2 acetyl groups
More lipophilic than morphine due to acetyl groups through acetic anhydride synthesis. → better BBB penetration (pharmokinetics improvement!)
50x more potent than morphine itself because more of it can enter the brain (bioavailability)
Heroin is NOT psychoactive in the brain: needs to be metabolized back into morphine in the brain to exert central effects (“prodrugs”)
Dilaudid (hospital heroin)
Semi-synthetic opioids: Codeine derived
Hydrocodone: e.g. Vicodin (analgesic); used as an antiussive (cough suppressant)
Semi-synthetic opioids: Thebaine derived
Antagonists of opioid receptors: naltrexone, naloxone (narcan)
Oxycodone: Percodan (oxycodone + aspirin), Percoset (oxycodone + acetaminophen)
Controlled-release oxycodone: e.g. OxyContin.
Buprenorphine: e.g. suboxone
Synthetic opioids
Methadone: used in treatment of opioid use disorder → opioid receptor agonist.
Fentanyl: 30-50x more potent than heroin, 100x more potent than morphine.
2 mg of fentanyl is a lethal dose for most people.
Carfentanyl: 10000x more potent than morphine → animal tranquilizer & anesthetic.
Sources of opioids: Global
Traditionally, licit opioids tend to come from India
Illicit opioids (e.g. heroin):
Past: Golden Triangle dominated illicit heroin trade ⇒ Southeast Asia (Northern Laos, Thailand, and Myanmar)
Now constitutes 1/3 of illicit heroin
Currently: Afghanistan now leads in illicit heroin trade (⅔)
Designer drug
Modified versions of controlled substances ⇒ Functional/structural analog of another drug
Improved potency, but variable and uncertain dosing
Technically unscheduled drug → not subject to laws.
Easier to make and cheaper.
Designer drug: Examples
Krokodil: derived from codeine → has active ingredient desomorphine
Powerful analgesic; “Russian heroin.”
Created because Afghan heroin decreased (due to US war)
Amateur chemistry used to make it results in toxic byproducts (HCl, red phosphorus, etc.) = scaly, gangrenous wounds, rotting of extremities.
Late 70s & 80s: Inadvertent by-product of home-synthesized drug (heroin analog) caused Parkinson-like effects in users (“frozen addicts”)
Cause: Drug manufactured contained impurity that metabolized into neurotoxin that kills dopamine neurons.
History of Opium: Ancient Use
Sumerians of Mesopotamia (4000 BC): valued the poppy plant (hul gil) → orally used for medical, analgesic effects.
China: the beginning of recreational use after >7000 years of medicinal use.
Origins in Asia and later Mediterranean regions
Traders brought opium to China in 9th century
Smoking opium created addictions fueled by British-owned opium from India
Led to the Opium Wars between China and Britain (1830-1860s).
History of Opium: Introduction to Europe & the US
Merchants of Venice brought opium to the West → widespread in mid-1800s.
Advertised as “cures”; causes pipe dreams
Available by mail order, grocery stores, etc. => e.g. Laudanum: 10% heroin dissolved in alcohol → cough suppressant, analgesic.
1803: Morphine identified as the principle active ingredient of opium
History of Opium: Heroin
End of 19th century: heroin introduced by Bayer Company in Germany
Marketed as cough suppressant lacking dependence-producing properties of morphine.
Banned in 1924 → illegal for doctors to prescribe heroin.
Misuse potential only fully understood beginning of 20th century → by 1900, 250,000 opioid dependent ppl.
Harrison Narcotics Act!
History: Contemporary Use of Opioids
1960s: Hippie!
Vietnam War: soldiers exposed to high-grade opioid products
Greater public acceptance of drug use and experimentation
1970-present: analgesic opioids have become one of the most highly prescribed of all drugs.
For a long time, prescription opioids (e.g. OxyContin) were prescribed for even minor things (e.g. headaches) ⇒ peak 2012
2019: ~75% of the 100,000 deaths in 2019 were due to opioids
Opium: Routes of Administration
Morphine is a weak base (ph ~8.5) → poorly absorbed in GI tract
Typically given IV in the hospital
Oral route less effective than IV: not rapidly absorbed from the digestive system → effects are slowed but prolonged (steady state levels)
Heroin:
Parenteral: IV → fall into disfavor: greater awareness of dirty needles/blood-borned pathogens/disease transmission
Snuff (intranasal)
Smoked
Opium: Absorption & Distribution
Heroine passes BBB easily
Heroin and codeine metabolized into morphine to act on opioid receptors.
Morphine taken orally → only ~15% get into the brain ⇒ significant first-pass metabolism by CYP3A4 enzymes (liver & gut)
HOWEVER: Codeine has ~90% bioavailability even if taken orally ⇒ dependent on lipophilicity.
Oxycodone and methadone have high bioavailability w/ oral administration.
Many opioids are bound to blood proteins → extends half lives.
Methadone has a half-life of 10-25 hours in bound form
Opium: Metabolism & excretion
Morphine half-life: ~2 hours
~90% of morphine is metabolized for elimination in urine and feces via concentration in bile
~10% of morphine is excreted in the urine unchanged.
Type of opioid receptors
All of these receptors are GPCRs (metabotropic) ⇒ slower, stronger signal due to signal amplification.
Mu:
Kappa
Delta
ORL1 (nociception receptors)
Locations of opioid receptors
Opioid receptors found throughout the brain but concentrated in the midbrain (periaqueductal gray: pain modulation), thalamus, hippocampus, striatum, and olfactory bulb.
Mu receptor widely distributed in brain + spinal cord → most important for analgesia and reward/euphoria.
Functions of delta and kappa receptors are widely unknown → occur in overlapping brain regions w/ mu receptor.
Principle effects of opioids:
Analgesia (periaqueductal gray, spinal cord)
Euphoria/reinforcement (VTA/NAc)
Vital life functions: acting on brainstem regions → breathing, vomiting, coughing
Opioids & Mu-Receptors
Opioids have inhibitory effects on neurons = CNS depressants
Post-synaptic: receptors activate a variety of K+ channels (GIRKS) = efflux of K+ = hyperpolarize the neurons & release firing → opioids are inhibitory NT
Pre-synaptic: receptors inhibit voltage-gated Ca2+ channels and activate K+ channels ⇒ decrease in NT release (glutamate, GABA, dopamine & acetylcholine) → opioids are inhibitory neuromodulators
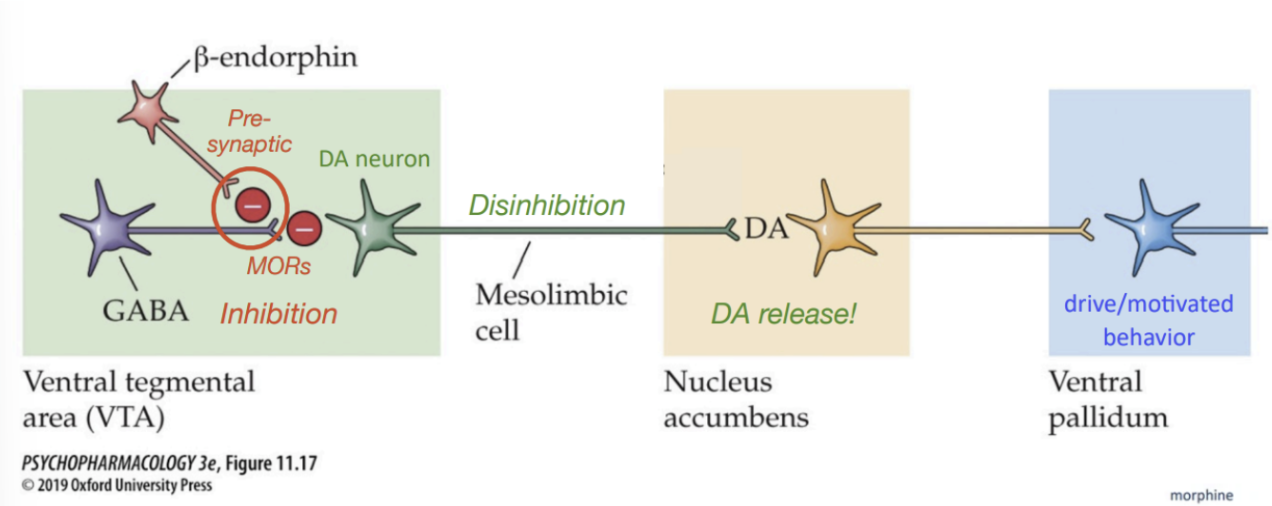
Effect of Opioids on Dopamine Reinforcements
Beta-endorphins (& exogenous opioids) bind presynaptic MORs on GABA neurons → inhibit Ca2+ channels
This block GABA release onto DA neurons in VTA → disinhibition of DA neurons.
Results in increased DA release in NAc (reinforcement)
Endogenous opioid neuropeptides
Enkephalins “in the head” (1975)
Greatest affinity for delta receptor
First endogenous opioid discovered.
Endorphins (“morphine within”)
Originally isolated in 1960s from camel pituitary glands
Put in storage, but when enkephalins were identified, he realized what he had found.
Greatest affinity for mu receptor
Dynorphins: binds to kappa receptor
Nociceptin: binds to ORL1 receptor
Endomorphins: binds to mu receptor
Competitive Mu Opioid Receptor Agonists
Morphine, methadone, LAAM (completely synthetic), Fentanyl, etc.
Mild-to-strong binding affinity to mu receptors depends on opioid ⇒ heroin & codeine are converted back to morphine in the brain, because they cannot bind to mu themselves.
Short-acting forms: powerful opioid “rush” (e.g. heroin, morphine, fentanyl)
Long-acting forms: weak opioid “rush” (e.g. codeine, methadone, LAAM) but lasts longer.
Competitive Mu Opioid Receptor Agonists: Primary Effects
VLF/respiratory depression
CNS effects:
Analgesia
Euphoria/reinforcement: due to enhances dopamine release in NAc
Pain components
Sensory component
Thermoceptive (heat or cold)
Mechanical (physical damage)
Visceral (organ damage)
Detection of pain involves circuits in spinal cord + brain (periaqueductal gray) w/ endogenous opioids.
Injury (nociceptive signal) → ascend pain pathway → somatosensory cortex = produces sensation of pain.
Descending projections: Opioids activate neurons in periaqueductal gray (midbrain) → inhibit pain perception neurons → less firing = weaker pain signal
Emotional component: aversive aspect of pain → opioids (limbic system, extended amygdala) blunts these aversive emotions.
Competitive Mu Opioid Receptor Agonists: Side Effects
Inhibition of brainstem effectors controlling VLF: Powerful respiratory depression at higher doses → lead to death
Nausea & vomitting (emesis trigger zone)
Constipation → treated with laxatives
With repeated use = powerful tolerance and physical & psychological dependence.
Competitive Mu Opioid Receptor Antagonists: Function
Derived from thebain and have no intrinsic efficacy on their own
Function: compete for opioid binding sites on MORs and prevent other opioids from binding.
Competitive Mu Opioid Receptor Antagonists
Naloxone:
High MOR binding affinity
Short half-life & rapid onset when given IV or intranasally
Treat/reverse opioid overdose
Naltrexone:
Longer duration or action and slower onset (oral)
Curbs euphoric effects of drugs = block MORs in hedonic hotspots
Treat substance use disorders like OUD
Effects of opioid: physiological
Nausea and vomiting: activation of MORs in brainstem regions controlling emesis.
Constipation: due to activation of opioid receptors in gut (inhibitory) = impedes motility ⇒ distension and delays emptying.
Profuse sweating
Pupilary constriction ⇒ Pinpoint pupils (sign of OD, parasympathetic response)
Sleep/sedation: produces drowsiness and lethargy, nodding off, “pipe dreams”
Long-term use can cause insomnia
Effects of opioid: cognitive performance & mood
Associated with inattention, difficulty concentration, perceptual distortions, memory deficits and executive dysfunction.
Calming effect, reduced anxiety, trance-like (feelings of detachment)
Reduced emotional rxn to pain
Intense momentary feeling of euphoria (associated with recreational use of strong opioids)
Opioid Overdose
Opioids are potent ligands → bind to MORs in areas of the brain like brainstem.
High doses/potent opioids: Comatose state with pinpoint pupils & severe respiratory depression.
Opioid analgesic overdose is the leading drug poisoning in the US
High level of overdose among heroin users:
Behavioral tolerance & conditioned environment
Mixing of drugs (e.g. fentanyls)
Reuse after a period of abstinence:
PD tolerance (amount of MORs) develops with regular drug use and recovers during abstinence ⇒ past dose becomes way too high & body cannot adjust.
Opioid Use Disorder
DSM-5 Diagnostic Criteria: 11 criteria related to impaired control, social impairment, risky use, neuropharmacological changes.
Risk factors ⇒ genetic components, young age, history of criminality or depression/anxiety, regular contact with high-risk people or environments.
Opioid withdrawal
Not as dangerous as alcohol or barbiturates
Onset ~6-12 hours from last dose → becomes less severe over time (can disappear within a week)
Restlessness and agitation
Yawning
Chills and sweats: Hyperthermia
Panting: short, jerky breaths
Goose bumps
Stomach, back & leg cramps
Vomiting and diarrhea
Twitching of diarrhea
Opioid Pharmacotherapies: Short-term
Short-term detoxification involving pharmacological intervention → antiquated, “legacy” treatment
Abrupt: antagonist of MORs injected into the body and leads to quick/severe onset of withdrawal symptoms (e.g. naltrexone)
Very unpleasant, can be dangerous
Opioid Pharmacotherapies: Long-term (Methadone)
Addressing craving and physical dependence factors in body
Methadone: fully synthetic opioid and full MOR agonist (high affinity, strong activation)
Less addictive than heroin (taken orally as pill or liquid)
Long half-life: 1 dose given under medical supervision lasts all day
People may become dependent on methadone and use it for prolonged periods of time.
Significantly reduces withdrawal symptoms + cravings for other opioids.
Reduces exposure to dirty needles/blood-borne diseases (e.g. hepatitis or HIV)
Opioid Pharmacotherapies: Long-term (Buprenorphine)
Buprenorphine: semisynthetic, partial mu opioid agonist → binds strongly but has weak effect (low efficacy)
Mixed agonist that can affect delta and K receptors as well
Downside: People still experience withdrawal symptoms + cravings when taking this opioid.
Misuse potential if crushed to take through IV or snuff
Suboxone: sublingual form ⇒ has buprenorphine (partial agonist) + antagonist naloxone (antagonist)
Taken as pill (correct way):
Naloxone is not well absorbed in the mouth
Buprenorphine is well-absorbed (weak base)
If tablets crushed and snorted/injected = naloxone bioavailability is high ⇒ blocks euphoric effect of buprenophine + lead to withdrawal.
Schizophrenia
“Split mind”: disconnect between thought, emotion, and behavior → different aspects of the same personality
With psychotic disorders, people experience psychoses ⇒ disturbances in thoughts & perception = results in loss of touch with reality
Treatment: behavioral therapy + use of antipscyhotic medications.
Positive Symptoms of Schizophrenia
Things that are present that shouldn’t be
Hallucinations: False sensory perceptions → auditory (hearing voices), visual.
Delusions: False, unshakeable beliefs held by individual.
Persecution
Grandeur
Control: belief that thoughts and behaviors are controlled by external forces.
Negative Symptoms of Schizophrenia
Things that should be present but are not
Difficulty expressing emotions and planning
Poverty of speech
Lack of motivation, anhedonia
Isolating, social withdrawal
Disinterest in day-to-day life, flattened emotional response.
Cognitive & Additional Symptoms of Schizophrenia
Cognitive:
Related to PFC:
Difficulty in attention and applying information to make decisions.
Poor abstract thinking, poor problem-solving.
Deficits in learning + memory
Low psychomotor speed
Sometimes, patients may be catatonic ⇒ may exhibit odd posing + remain in catatonic positions for hours.
DSM-5 Criteria for Schizophrenia
Two or more symptoms must be present for at least 1 month → at least one must be either 1/2/3
Hallucinations
Delusions
Disorganized speech
Grossly disorganized or catatonic behavior
Negative symptoms (e.g. affective flattening, avolition, alogia: lack of speech)
Continuous disturbance for 6 months
Social and/or occupational dysfunction for a significant portion of time.
Onset of schizophrenia
Typically in early adulthood (~20.5 years old) → related to PFC development
Varies based on biological sex: more males experience symptoms in earlier years (16-35), more women than men experience first episode after age 36.
Schizophrenia: Hereditary
Risk increases with the number of shared genes (relatedness)
Genetic analysis found genes involved in processes like neuronal migration, differentiation, and growth.
48% of concordance in MZ twins → 52% to environment
Children of 2 parents with schiz: 46%
Schizophrenia: Environmental
Maternal environment: mothers who are sick during pregnancy = inflammation that affects children’s neuronal development.
Birth complication: Excessive cortisol release
Brain abnormalities in schizophrenia: Structural changes
Cortical grey matter loss (cerebral atrophy): excessive synaptic pruning in perinatal & adolescent periods.
During adolescence:
Average annual rate of cortical gray matter loss in adolescents w/ schizophrenia is >/= 2x as much as healthy adolescents.
Pathologically exaggerated levels of pruning in temporal, parietal lobe, frontal cortex.
Ventricular enlargement due to gray matter (brain volume) loss.
Ventricles are filled with cerebrospinal fluid
Brain abnormalities in schizophrenia: Structural changes (p2)
Disorganized hippocampal cytoarchitecture: neuron migration issues in development.
Post-mortem immunohistochemistry images show a haphazard arrangement of pyramidal cells in the hippocampus of patients with schizophrenia (vs. normal, organized, parallel, laminar structure)
If neurons are not in the right place = do not make appropriate synaptic connections.
Shrinking of dendritic trees leading to connective failures: due to issues with synaptogenesis.
Etiology of schizophrenia: Two-hit model (First Hit)
Early stage “first hit”: perinatal period
Genetic predisposition & gene expression + environmental insults (viruses, toxins, poor nutrition, birth complications, activated immune system) → neurodevelopmental abnormalities (neuron formation, migration, synaptogensis, pruning, apoptosis.)
Etiology of schizophrenia: Two-hit model (Early signs)
Latent stage: first few years of life
Early subtle signs: motor abnormalities, apathy, social withdrawal, attentional and information-processing deficits.
Etiology of schizophrenia: Two-hit model (Second hit)
Excessive synaptic pruning (abnormal neuronal connectivity & function) + later environmental insults (stress, substance use, HPA dysfunction) → greater impairment of cognitive functions, worsening of negative symptoms, development of positive symptoms.
Neurodevelopment/hypofrontality hypothesis
Defects in early PFC development result in reduced PFC activity
Evidence:
Less frontal cortex activation in people w/ schizophrenia compared to unaffected twin at rest & during cognitive tasks
Appears to be a loss of glutamatergic neurons in particular.
Dopamine hypothesis: Schizophrenia
Schizophrenia due to hyperactivation of dopamine circuits → Increased salience to environmental cues
Evidence:
Drugs that enhance DA activity can produce positive symptoms of schizophrenia (e.g. monoamine psychosis)
First effective anti-psychotic drugs are dopamine D2 receptor antagonists.
Contrasting:
HOWEVER, antipsychotic drugs that block D2 receptors take several weeks to work & do not improve negative and cognitive symptoms of schizophrenia.
Glutamate hypothesis: Schizophrenia
Schizophrenia due to hypoactivation of glutamate circuits (too little GLU signaling)
Evidence:
Drugs that mimic some symptoms of schizophrenia are glutamate NMDA receptor antagonists (e.g. PCP, ketamine block allosteric site = non-competitive)
However, inconsistent findings concerning brain glutamate lvls of ppl with schizophrenia and NMDAR modulators have not shown consistent efficacy in treating schizophrenia.
Hypothesis for Schizophrenia: Mesocorticolimbic system dysregulation
Neurodevelopmental issues leads to loss of PFC glutamatergic projection neurons ⇒ mesocorticolimbic system dysregulation.
Reduced glutamate activity in PFC = weakened mesocortical dopamine activation (negative/cognitive symptoms)
Normally: Excitatory glutamate neuron from PFC synapses to dopamine neurons in VTA w/ projections to PFC
Less glutamate neuron in PFC = less dopamine release from VTA → PFC.
Enhanced mesolimbic dopamine activation (activation)
Glutamate neurons synapse on GABA interneuron in VTA → less GLU neurons to excite GABA = leads to disinhibition of dopamine neurons that project to NAc.
History of antipscyhotic medications
Prior to mid-1950s: no effective treatments → frontal lobotomies (separation of frontal lobe from rest of brain) ⇒ only made patients more docile, came at cost of executive function & personality.
First was chloropromazine (trade name Thorazine)
Dopamine D2 receptor antagonist ⇒ reducing positive symptoms of schizophrenia.
Led to the closure of many psychiatric hospitals in the U.S. ⇒ Thorazine revolution
First generation “typical” antipsychotics
Developed before 1975
Examples: chloropromazine, haloperidol, etc. ⇒ primarily dopamine metabotropic D2 receptor antagonists.
Higher D2R affinity = lower effective dose required.
Effective in treating positive symptoms of psychosis, but less effective in treating negative or cognitive symptoms.
May cause serious motor side effects ⇒ in 40% of users.
“Extrapyramidal” motor side effects = linked with nigrostriatal motor system issues (e.g. basal ganglia) NOT motor cortex.
Acute dystonia (spasms of muscles), akathisia (motor restlessness), Parkinsonism (rigidity, tremor), tardive dyskinesia (oral-facial tics)
Second-generation “atypical” antipsychotics
Recently developed (1980s-current)
Clozapine, ziprasidone
Weak affinity to dopamine D2 receptors ⇒ fewer adverse EPS side effects.
Antagonism of dopamine D3 & D4s (lower concentration in basal ganglia) and serotonin 2A receptors (high affinity)
Serotonin modulation (in PFC) = improve mood and reduce risk of EPS ⇒ modulate DA motor pathways.
Atypicals can alleviate positive, negative, and cognitive symptoms of schizophrenia when they work w/o motor side effects.
Issues with atypicals
Issues:
Only ⅓ of individuals respond well to atypical antipsychotics.
Unwanted metabolic side effects ⇒ weight gain (5-10% bodyweight gain on average), sedation, and more.
Third-generation Dopamine System Stabilizers (DSS)
Aripiprazole (trade name Abilify) was approved by FDA in 2002
Competitive DA receptor partial agonist → modulating DA activity at D2, D3 and D4 receptor subtypes.
High affinity for DRs, but activates receptor to a lesser degree (reduced efficacy) compared to dopamine.
Function: stabilizes regional DA activity
In mesolimbic system where DA activity is too high: weakly activates DA receptors & prevents endogenous dopamine from binding ⇒ reduces positive symptoms.
In mesocortical pathway = mildly enhances DA signaling ⇒ reduces negative & cognitive symptoms.
DSS side effects
Motor restlessness (akathisia), though with greater tolerability.
Minor weight gain
Insomnia
Nausea
GI issues
Limitations: not as effective with severe schizophrenia
Administration: Anti-psychotics
Taken orally or slowly dissolving depot injection (can last up to a month)
Effects of antipsychotics are not accelerated by IV injection because effects typically take several days/weeks to develop (metabotropic, can influence gene expression)
PK: Antipsychotics
Absorption & distribution:
Lipophilic are lipophilic and readily absorbed from the digestive tract and distributed throughout the body, easily crossing bloodbrain and placental barriers
Often absorbed in body fat and released slowly
Metabolism & elimination:
Cytochrome P450 enzymes
Long halflives (11-58 hours, particularly for typicals)
Physical Effects of Antipsychotics: Neuroleptic EPS
First-generation “typical” antipsychotics cause neuroleptic (motor) EPS in 40% of patients → resemble symptoms of Parkinson’s disease:
Dulled facial expression, muscle rigidity and tremor in the limbs, slowing or loss of coordinated movement, weakness in the extremities, etc.
Tardive dyskinesia—involuntary tic-like repetitive movements of the face that can occur dozens of times per minute
Other physical effects of antipsychotics
Temperature regulation (easily influenced by changes in the environment), weight gain, changes in cardiac function, and blood pressure
Some first- and second-generation antipsychotics increase prolactin levels, which can lead to:
Biological females: menstrual irregularities, milk production, infertility, decreased libido
Biological males: gynecomastia, erectile dysfunction, low T / libido, infertility
Subjective effects of antipsychotics
Particularly for first and second generation
Tiredness
Slowed and confused thinking
Trouble concentrating
Anxiety and irritability
Internal restlessness, but physical restraint due to movement difficulties
Results in difficulty with patient compliance
Antipsychotics do not result in significant mesolimbic DA release, and are not reinforcing.
Performance effects of antipsychotics
Inconclusive effects on cognitive performance overall
Sedating effects
Reduced sexual interest and performance (low libido)
Possible abnormal menstrual cycles and infertility in women; low T in men
Tolerance, Lethality, Withdrawal: antipsychotics
Tolerance:
Tolerance doesn’t develop → therapeutic dose often maintained for years without decrease in effectiveness.
Extremely safe with high TI (~100-1000); Practically impossible to overdose
Withdrawal:
Physical dependence is rare or very mild
Possible exaggeration of psychotic symptoms, headache, and nausea when drug is withdrawn but fairly uncommon.
Cannabinoids: Legal designations
Cannabis: “umbrella” term ⇒ refers to plant genus (C. sativa and C. indica)
Hemp: cannabis plant with =/<0.3% THC → used for strong fibers
Marijuana: cannabis plant w/ >0.3% THC
C. sativa
Hemp plant: rope, linen, paper from stalks → Hemp has higher CBD content than marijuana.
Male plants usually used for hemp or breeding.
Marijuana is C. sativa with >0.3 THC
Cannabinoid Source
Cannabinoids found in leaves (low THC) and female flowers (high THC)
Cannabis buds: form appendages called “trichomes” that secrete droplets of “resin” → composed of cannabinoids (fatty acids = oil texture)
Female plants have larger cannabis bud/flower! → typically cultivated for resin.
THC (psychoactive) and >100 other cannabinoids like cannabidiol (CBD; not psychoactive)
Cannabis indica
Grown in India and surrounding regions
Usually higher levels of CBD and lower THC → however, nowadays, cultivated strains offer higher THC.
Uses of cannabis
Leaves (low THC) and buds (high THC) are smoked or psychoactive components of resin can be ingested (“edibles”)
Pollen-collecting resin from female plant:
Produced by plant to capture pollen + protect seeds
Dried: hashish → concentrated resin from plants
Hash oil: resin boiled in alcohol and then filtered ⇒ concentrates the psychoactive chemicals.
Sinsemilla
Sinsemilla female plants: unfertilized female plant produces flowers but not seeds → greatest resin producers.
Growing female plants away from male plants → reproductive overdrive to attract pollen (even though there are none) ⇒ increased bud growth.
History: Early cannabis use
Stone Age/Upper Neolithic: Archeological evidence from Taiwan of hemp used to make rope & cloth.
2350 BCE: Egyptian Pyramid Texts carved into pyramids mention plant that made rope + treating glaucoma and inflammation.
Hair samples taken from Egyptian mummies indicate higher levels of cannabis use.
Over 12,000 years ago: Cannabis may have originated in Central Asia → first cultivated for fiber in China and India
Medical use in China; religious use in India.
1000 CE: Hashish use in Arab world.
History (Cannabis): American & European use
Mid-1600s: Colonia; America Cannabis (HEMP) Use
Colonies produced rope, cloth, canvas, sacks and paper from hemp leading up to Revolutionary War
1800s: European recreational cannabis use
French troops returning from Egypt brought hashish home (1800) → began to be imported
Club des Hachichins (1846): a book club of high society artists → revelers consumed hashish and wrote about experiences.
Most were not aware of association between hemp and hashish.
England: Traces of cannabis found in Shakespeare’s pipes.
History of Cannabis Use: 20th century
U.S. Recreational Cannabis Use & Subsequent Propaganda
Term “marihuana” was promoted in 1930s as a racist attempt to demonize its use = link to immigrants from Mexico
Reefer Madness: U.S. propaganda film → Cannabis as “Devil’s weed” ⇒ use leads to unlawfulness/death/destruction.
Nixon’s “War on Drugs”: Cannabis as a “gateway drug”
→ Propaganda = regulations = criminalized possession.
U.S. Cannabis Legislation
1850: first listed in US Pharmacopoeia (USP)
1910: “Recreational” use for psychoactive effects begins to emerge in the U.S.
1937: Marijuana Tax Act → not made illegal, but high taxes imposed on use and distribution.
1942: Removed from USP → claims that there is no medical use.
1970: Controlled Substances Act
Tax Act repealed but marijuana said to have no medical use.
Beginning mid-1970s ⇒ >12 states decriminalized possession of small amounts of cannbis + legal medical use in California.
Phytocannabinoids
THC: psychoactive, analgesic (nerve pain, etc.), anti-emetic (anti-convulsant), and appetite-stimulating effects.
Partial agonist of CB1
Isolated from cannabis in 1964 in Israel
Primary psychoactive compound
Content higher in the resins (hashish)
Selection for this compound = 10x increase in THC content since 1960
Other: Cannabidiol (CBD) and cannabinol (CBN)
Extracted phytocannabinoids: Sativex
Sativex: whole plant medicinal cannabis extract & sprayed under the tongue.
Developed to treat multiple sclerosis and neuropathic pain
Not FDA approved (available in UK)
1:1 radio of THC: CBD
Extracted Phytocannabinoids: Epidiolex
Epidiolex:
CBD
Treats seizures in children ⇒ FDA approved for severe forms of epilepsy (Lennox-Gastaut syndrome, Dravet syndrom) in children aged 2+
First FDA approved drug w/ cannabis derivative.
Synthocannabinoids
Recreational cannabinoid-like chemicals ⇒ “designer”
Most are produced in China
Unregulated in U.S. market initially in early 2000s → initially sold as herbal incense ( => synthocannabinoids sprayed onto plants)
2012 Synthetic Drug Abuse Prevention Act ⇒ placed in Schedule I
Can contain many other drugs (including hallucinogens like PCP), methamphetamine-like compounds, and toxic products (rat poison, embalming liquid)
Effects: 40-100x more potent than THC
“High”
Produce extreme anxiety, confusion
Psychotic effects ⇒ e.g. paranoia and hallucinations.
Endocannabinoids
Cannabinoid receptor agonists synthesized by the body. => Fatty acids synthesized from phospholipids in the cell membrane
Arachidonoyl ethanolamide (anandamide)
Partial-agonist of CB1 → lower efficacy (ceiling effects)
Discovered in 1994 (“internal bliss”)
2-arachidonoylglycerol (2-AG)
Full agonist of CB1 → higher efficacy
Mechanisms of endocannabinoids
Retrograde messengers
Are released from post-synaptic cell to active presynaptic CB1 receptors
Produce complex effects in the brain
Not a classic neurotransmitter ⇒ modulates the NT release presynaptically (homeostatic property)
Example:
One neuron fires too often = excessive excitation of postsynaptic neuron
Activation of homeostatic property in post- neuron = synthesizes and releases endocannabinoids.
Binds to receptors on pre- ⇒ inhibition + regulate the activity of presynaptic neuron
Cannabinoid receptor: CB1
First identified in 1988 and occur mainly in the brain and less in PNS.
Endocannabinoid effects of presynaptic receptors:
CB1 are metabotropic (like MORs) → work via G-proteins to:
Inhibit Ca2+ channels
Open K+ channels
→ Overall inhibitory
Found on axon terminals of most GABA-ergic neurons, but also on other neurons (e.g. NE, DA, 5HT)
Work as “on-demand” signal ⇒ transient but HIGHLY SPECIFIC signaling between neurons.
Effects of cannabinoids on CB1 in different brain areas:
Amygdala: stress, anxiety ⇒ relaxation.
Ventral striatum: euphoria, reinforcement
Dorsal striatum + ventral striatum: motor coordination ⇒ cannabinoids = issues.
Cerebral cortex ⇒ effects: altered cognition, memory disruption, sensory/perceptual disturbances.
Periaqueductal gray: involved in pain modulation ⇒ cannabinoids as analgesics
Hypothalamus: direct influence on appetite ⇒ stimulation
THC effects on CB1
Partial CB1 receptor agonist → milder “ceiling” effects, psychoactive effects plateau
Activates CB1 receptors non-selectively (floods entire brain) ⇒ NOT a retrograde NT.
Disrupts temporal precision of natural signaling
Global changes in sensory, memory, motor, cognitive & reward processing.
Cannabinoid Receptors: CB2
Occur in immune system, and other tissue (bone, adipose cells, GI tract) ⇒ can also be found in the brain, but less.
Mechanism of Cannabinoid-Induced DA Release
Stimulation of CB1 by THC on GABA terminals → reduce GABA amount released on DA projections.
Disinhibition of DA → increased DA release in NAc → reinforcing effects of THC.
→ acute increases in DA release elicited by THC are relatively small compared to other psychoactive substances.
Cannabinoid: Absorption (Oral)
Cannabinoids are WEAK ACIDS & highly lipid-soluble.
Oral administration (“edibles”)
Un-ionized in the gut. → Good, slow, steady absorption from stomach/intestine
Significant first-pass metabolism
THC is converted into fat due to lipophilic properties.
Peak effects: 1-3 hours after ingestion.
Cannabinoids Absorption: Inhalation
Inhalation (smoke or vapor)
THC peaks in blood by 6-10 minutes
Peak behavioral effects: 30-60 minutes
Absorption increased by depth of inhalation NOT duration of holding the smoke in.
Time course:
Blood THC levels decline rapidly after smoking cannabis
Falls to ~60% by 15 min, ~20% by 30 minutes
Complete elimination is slow ⇒ persistence in fat tissues (>2 weeks)
Cannabinoids: Distribution
All areas of the body (blood flow)
Bioavailability: 10-35% via inhalation; 4-12% via ingestion
Blood half-life: 1-3 days (occasional use); 5-12 days (chronic use)
THC remains in body fat for many weeks.
Subjective intensity of “high”:
IV + Smoked: intense early on, then decreases over time rapidly.
Oral: slower onset, longer duration, overall rated “high” is lower.
Cannabinoids: Metabolism & excretion
Starts as soon as cannabinoids enter the body
Delta 9-THC converted into 11-hydroxy-delta 9-THC ⇒ more active/lipophilic than THC, crosses BBB more easily, a full agonist of CB1.
Creates ~100 mostly inactive metabolites.
Half-life of cannabinoids varies considerably between individuals ⇒ affected by dose, frequency of use, etc.
Full elimination of cannabinoids from the body ⇒ 12*6 (half-lives) = 72 days in chronic users.
Acute Physiological Effects: Cannabis
Cardiac acceleration, dry mouth, reddening of eyes (vasodilation)
Drooping of eyelidss are common (muscle relaxation → CB1 receptor signaling) = “stoned”
May cause drowsiness + increased sleep time.
Higher doses can interfere with sleep
Often causes hyperphagia (“munchies”): cannabinoids interact with appetite control neurons in hypothalamus
Cannabinoid signaling interfere w/ NPY neurons (that typically inhibits appetite) → disinhibition leads to “hunger” signaling.
Acute Psychological/Subjective Effects: Cannabis
Mood: Euphoria, relaxation, giddiness, and variations in mood (typically elevated mood/excessive cheerfulness & contagious laughter)
Some people can report anxiety + adverse effects (paranoia or depression)
Dose-dependent: larger doses ⇒ increased likelihood of adverse effects (disinhibition of circuits in the amygdala for example)
Strain of cannabis & THC:CBD ratio
CBD: antipsychotic properties, calming effects ⇒ low CBD levels + high THC levels = increase likelihood of paranoia/depression.
Individual differences:
Genes can influence response
Personality
Psychological states
Acute Subjective Perceptual Effects: Cannabis
Sensory alterations due to THC’s effects on CB1s:
Affect areas of sensory processing: sensory cortices like inferior colliculus and posterior thalamus.
Leads to incongruencies (things don’t look/sound the way it should) = funny.
Temporal disintegration: likely involves cerebellum (usually fine-tune rhythmic activity = estimate time) ⇒ high = time feels elongated.
Others: intensification of colors, blending of patterns & objects.
Cognitive impairments: Cannabis
Cognitive impairments in attention + memory (STM formation & removal); interference with visuomotor skills (e.g. attention needed to drive)
Unlike alcohol, cannabis users report awareness of driving impairments
Memory:
No effects on recall of well-learned or recognition memory.
Disrupt short-term memory:
Recall of words or narrative material
Temporal disintegration:
Loss of ability to “keep things in mind”
Users often report disjointed conversations.