UNIT 4 CLS 292 ANEMIAS STUDY GUIDE
1/114
Earn XP
Description and Tags
finished
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
115 Terms
what is anemia? (learning objective)
most common hematologic disorder and is a reduction below normal in hemoglobin, hematocrit, or red blood cell number that is normally found in individual according to age and sex or premature RBC destruction
anemia is a secondary condition
what are the general symptoms of anemia? (learning objective)
generally related to the underlying lack of O2-carrying capacity
fatigue
weakness
dizziness
tachycardia
pallor of skin and mucous membranes
what specific symptom would a patient with hemolytic anemia have? (learning objective)
jaundiced (due to accumulation of bilirubin)
what specific symptoms would a patient with iron-deficiency anemia have? (learning objective)
PICA
smooth tongue
spoon-shaped nails (koilonychia)
what specific symptom would a patient with megaloblastic anemia have? (learning objective)
big, beefy tongue
what are the effects of sudden anemia? (learning objective)
tissue hypoxia → causes EPO to increase
the bone marrow is stimulated for various reasons:
to increase stem cell activity → results in erythroid precursors
shortening of maturation time
premature release of retics → if the hypoxia is severe, it could release NRBCs
essentially the sudden effects of anemia causes an increased demand of RBCs which causes the normal maturation of RBCs will be disrupted
what is the BMs response in when there is sudden anemia?
there will be a presence of “shift” retics or stress retics
retics will usually lose their RNA within 24 hours in the PB but in stress retics, it will take 2-2.5 days to lose their RNA
stress retics are released earlier from the BM and will have increased RNA
increased RBC production (3-fold) in moderate anemia and (5-fold) in severe anemia
bone marrow is hyperactive with increased proliferation → erythroid hyperplasia
what determines the maturation of reticulocytes?
hematocrit → the lower the hematocrit, it will move out of the BM earlier and mature in the reticulocytes for a longer time
what is iron deficiency anemia? (learning objective)
occurs when the body storage of iron have been depleted which can occur due to inadequate dietary iron uptake, inadequate iron absorption, and abnormal iron loss
what is the normal distribution of iron?
60-80% of iron is functional and serves in metabolic (hemoglobin/myoglobin) or enzymatic functions
remainder of iron serves as a transport (uses transferrin to be transported) or storage forms (ferritin and hemosiderin → mostly in liver and bone marrow)
iron cannot exist alone and must be bound to a protein which is transferrin
how does iron get in and out of the body?
iron has a regulatory system which is controlled through the hormone, hepcidin→ iron will leave the body by being stored in the cells and getting sloughed off… when RBCs are degraded, macrophages will recycle them
what is the pathogenesis of iron-deficiency anemia? (learning objective)
blood loss (storage pool of RBCs is being depleted)
GI bleed, heavy menstruation, injury or accident
really bad diet (rare)
diet is multifaceted, meaning many things can be affecting diet
iron may not be absorbed
treated with oral iron supplements if anemia is not due to GI bleed and always establish the cause of IDA first
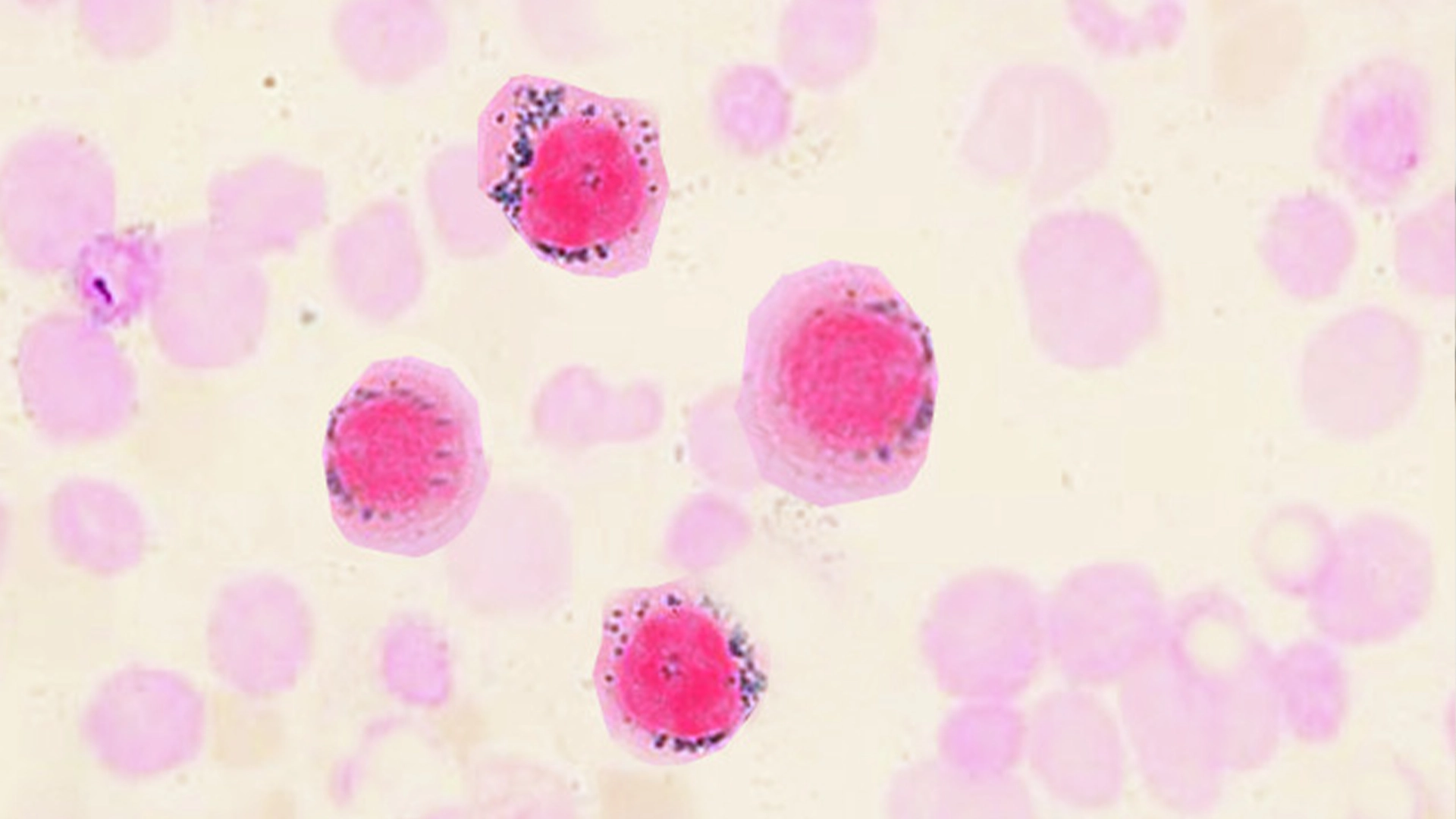
what would the RBC morphology look like in those with iron-deficiency anemia? (learning objective)
cells are hypochromic and microcytic with increased anisocytosis and poikilocytosis (elliptocytes)… reticulocytes are decreased and platelets are increased
anisocytosis is because each new wave of RBCs has less and less iron
lack of iron is the cause of the decreased reticulocytes
myeloid:erythroid ratio is decreased due to increased erythropoiesis
all RBC indices are decreased
what would the results of the iron studies look like in those with iron-deficiency anemia?
decreased serum iron
increased total iron binding capacity (TIBC)
TIBC → the total amount of Fe capable of being bound to transferrin
decreased ferritin
ferritin → primary storage form of soluble iron; readily released for heme synthesis and consists of a spherical protein shell that can store up to 4500 iron atoms
ferritin without its shell = apoferritin
what is the first stage of iron deficiency anemia?
iron depletion
iron stores are exhausted causing decreased ferritin
there is no anemia yet and RBC morphology is normal
RDW may be increased → could be the first hematologic indication of a developing iron deficiency anemia
what is the second stage of iron deficiency anemia?
iron deficient erythropoiesis
anemia and hypochromia is not detectable
RBCs may be slightly microcytic
will have decreased transferrin saturation and increased erythrocyte protoporphyrin
what is the third stage of iron deficiency anemia?
iron deficiency anemia
iron tests are markedly abnormal → severely deficient total body iron
microcytic hypochromic RBCs
what are the general characteristics of anemia caused by abnormal iron metabolism?
result of a block in the incorporation of iron into the protoporphyrin ring to form heme and defective iron utilization (usually due to the slow release iron from macrophages)
has a positive iron balance and could lead to an increase in iron stores (especially in spleen, liver, and BM) which results in increased serum ferritin levels
results in a lack of iron for Hb synthesis and a blood picture that is similar to IDA
iron is absorbed at normal rates but it is used at less than normal rates for erythropoiesis
what is sideroblastic anemia? (learning objective)
body has inefficient erythropoiesis due to a defective function of the HSC
elevated body iron storage, serum iron and serum ferritin is increased
decreased TIBC
low reticulocyte count and accumulation of iron in the mitochondria of the erythroid precursors
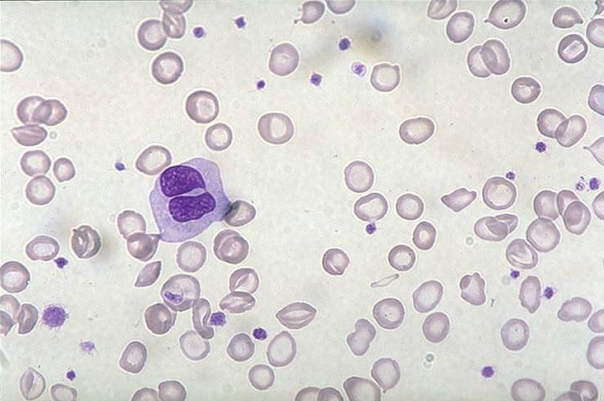
what would the PB look like in sideroblastic anemia? (learning objective)
ringed sideroblasts → siderotic granules surround the nucleus of the NRBCs due to the defective Hb synthesis
will have a dimorphic population of normochromic and hypochromic cells
MCV, MCH, and MCHC may be normal
RDW will be increased and has dual peaks on the histogram
will have a dual population of macro and microcytes
mild macrocytes are found in sideroblastic anemia that is associated with alcoholism
RBCs could have pappenheimer bodies
must use prussian blue to confirm sideroblastic anemia
what can cause sideroblastic anemia?
can be sex-linked recessive gene (hereditary sideroblastic anemia)
can be acquired and could be associated with other things like leukemia, drugs (can interfere with heme synthesis) and lead poisoning (basophilic stippling) (also benzenes)
more common
lead poisonings blocks enzymes in the heme biosynthetic pathway → can cause mental defects
would actually have siderocytes
usually moderate to severe anemia
what is the treatment for sideroblastic anemia?
pyridoxine therapy
usually tried on those who have hereditary sideroblastic anemia
less than ½ experience a return to normal Hb levels
iron overload will decrease responsiveness which is treated through phlebotomy and chelation therapy
folic acid
only given to those with megaloblastic features (seen in liver disease & alcoholism)
secondary hereditary anemia could be from a disease, toxin, or drug which is corrected through treatment of the primary disease or the elimination of toxin/drug
what are porphyrias?
a group of inherited disorders that are characterized by a block in porphyrin synthesis due to a defect in one or more enzymes in the pathway of heme synthesis → the heme precursors will accumulate in the tissues and large amounts are excreted in the urine (port wine) and/or feces
common lab findings → photosensitivity, abdominal pain, neuropathy
what is congenital erythropoietic porphyria (CEP)?
a rare autosomal recessive disease where excess porphyrins are deposited in body tissues and excreted in urine and feces → presence of CEP can be verified by intense fluorescence with UV light
cause of hemolytic anemia that is with CEP is unknown
what would the RBC morphology look like in those with congenital erythropoietic porphyria (CEP)?
mild to severe normocytic anemia with anisocytosis and poikilocytosis and significant polychromatophilia (reticulocytes) and NRBCs
RBCs will fluoresce with UV light
BM will show erythroid hyperplasia (more cells) and a portion of the normoblasts will have intense fluorescence
serum iron and storage is usually normal
large amounts of uroporphyrin I and coproporphyrin are excreted in urine and feces
what is erythropoietic photoporphyria (EPP)?
an autosomal dominant trait that causes a overproduction of protoporphyrin-immediate precursor of heme → ends up building up in the cell and leaks into the skin (causes hypersensitivity to sun) dermal capillaries and can be found in the skin, liver, blood, and feces (will not be present in the urine because of its insolubility)
adequate amounts of heme are produced → no anemia is present
what are the laboratory findings of erythropoietic photoporphyria?
cytoplasm of normoblasts fluoresces intensely but blood and BM will usually have no abnormalities
RBC’s plasma and feces contain large amounts of protoporphyrin which is not found in urine
what is the therapy for congenital erythropoietic porphyria?
quality of life is improved by minimizing the scarring and mutilation with effective dermatologic treatment so avoidance of sunlight is critical & blood transfusions with administration of chelators (to avoid iron overload)
most usually do not survive beyond 50
attempts to decrease excess porphyrins have been unsuccessful
what is the treatment for erythropoietic photoporphyria?
protect skin from sunlight (high does of B-carotene have been seen to improve tolerance to sunlight) and blood transfusions may be used to suppress erythropoiesis
splenectomy could be helpful if hemolysis and splenomegaly are prominent
this is because spleen would want to cull the abnormal cells (driver)
IRON IS TRAPPED IN MACROPHAGES
how are macrocytic anemias characterized? (learning objective)
abnormally large RBCs (MCV >100 fL) with normal hemoglobin content (MCHC) and can have megaloblastic (vitamin B12 or folic acid deficiency) and non-megaloblastic anemia that are caused by different things
non-megaloblastic anemia is usually a result of an acute blood loss resulting in a premature release of reticulocytes (shift retics)
associated with: alcoholism, hypothyroidism, liver disease, splenectomy, pregnancy
what is the pathogenesis of megaloblastic anemia? (learning objective)
vitamin B12 and/or folate deficiency (from bad diet, absorption problems, folate-antagonist drugs)
deficiency makes it hard to make DNA but RNA production runs smoothly
nucleus which is full of DNA lags behind the cytoplasm which is full of RNA in development and the cell divides more slowly
maturation is asynchronous (cytoplasm is mature but nucleus is immature) in the bone marrow
treatment depends on the cause of anemia
what would the RBC morphology look like in those with megaloblastic anemia? (learning objective)
red cells are macrocytic (would see great big oval macrocytes) with hypersegmented (>6) neutrophils → moderate-marked poik and aniso, NRBCs and inclusions like basophilic stippling and howell-jolly bodies
what is pernicious anemia? (learning objective)
a type of megaloblastic anemia that occurs when there is a vitamin b12 deficiency → gradually develops (sneaky) and has a defective cellular maturation due to impaired NA synthesis
intrinsic factor (IF) vitamin b12 complex (NEED IF-VITAMIN B12 COMPLEX) binds to receptor sites on epithelial cells of the small intestine and is absorbed in which B12 is then transported by protein to liver, BM, and other sites of rapidly dividing cells
in a lot of cases, vitamin b12 deficiency is secondary to a deficiency of intrinsic factor
what are the various clinical findings for pernicious anemia? (learning objective)
typical anemic symptoms → fatigue, weakness, pale or yellowish skin (due to bilirubin build up), irregular heartbeats (can also hear their heartbeat), shortness of breath, dizziness or lightheadedness, chest pain, cold hands and feet
smooth pale tongue or beefy red tongue
loss of weight and appetite
atrophy of gastric parietal cells
neurologic disturbances → leads to megaloblastic madness
altered personality, dementia, spastic weakness and ataxia due to demyelination of lateral and posterior columns of spinal cord
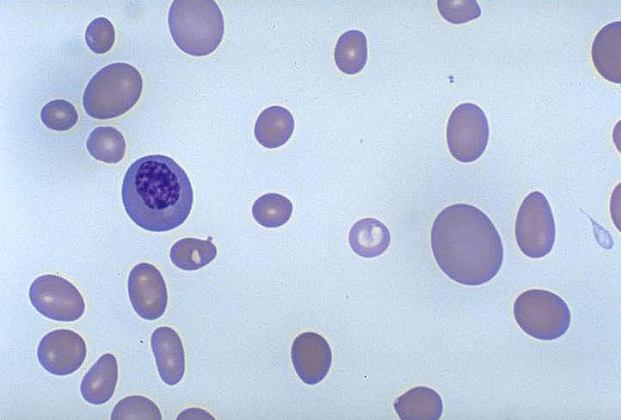
what would the RBC morphology look like in those with pernicious anemia? (learning objective)
macrocytic and tend to be elongated (ovalocytes) with an increased MCH and normal or slightly decreased MCHC
leukocyte and platelet development will be affected by being decreased
in BM, nuclear/cytoplasmic asynchrony is present so the end product is macrocytes → may see giant bands, metamyelocytes, and hypersegmented neutrophils (specific for megaloblastic anemia)
may see howell-jolly bodies and cabot rings
moderate to severe aniso and poik (usually more severe when anemia is severe)
polychromatophilia
what is used to determine the cause of pernicious anemia?
gastric analysis
determines cause of pernicious anemia which is a decrease in acidic gastric material secretions
schilling test
determines vitamin B12 malabsorption
radioactive-labeled vitamin B12 is taken orally then a 12 hr urine specimen is collected → labeled vitamin B12 is measured
about ~7.5% of the administered dose should be present in the urine specimen → if less, then the test is consistent with B12 deficiency
what is folic acid deficiency? (learning objective)
another type of megaloblastic anemia and is more common due to dietary deficiency and defective absorption with dietary deficiency being the most common (found in eggs, milk, fruit, and leafy vegetables)
major role is in DNA synthesis
body store in the liver is small and the minimum daily requirement is 50 micrograms → anemia will develop in 3-6 months in severe folate deficient diet
malabsorption syndromes can be caused by different intestinal disorders
certain types of anemias can involve rapid proliferation of HSC → can lead to folate deficiency
what is the therapy for megaloblastic anemia? (learning objective)
treat with specific deficient vitamin (anemia symptoms go away quickly after treatment) but will cause a rise in retics 4 days after therapy
degree of retics are proportional to the severity of the anemia
vitamin b12 therapy could reverse the peripheral neuropathy but spinal cord damage is irreversible
pernicious anemia has to be treated with lifelong monthly parenteral doses of b12 because of the inability to absorb oral vitamin b12
what is nonmegaloblastic macrocytic anemia? (learning objective)
has 3 common causes → liver disease, alcoholism, and reticulocytosis
macrocytes are not as pronounced and are usually round not oval
hypersegmented neutrophils are not present
leukocytes and platelets are normal
what is the most common disease associated with nonmegaloblastic macrocytic anemia?
liver disease → minor finding among the other abnormalities associated with liver disease
RBC survival seems to be significantly decreased in: alcoholic liver disease, infectious hepatitis, biliary cirrhosis, obstructive jaundice, and unknown reason (idiopathic)
anemia is usually mild → average Hb is 12 g/dL with normocytic, macrocytic or microcytic (may have a high RDW) and thrombocytopenia
how is alcoholism associated with nonmegaloblastic macrocytic anemia?
macrocytosis is probably the result of folate deficiency due to decreased dietary intake but alcohol seems to also interfere with folate metabolism and also with:
reticulocytosis is associated with hemolysis and GI bleeding (due to stress)
secondary to hepatic dysfunction → decreased in coagulation proteins or thrombocytopenia
hypersplenism from increased portal and splenic vein pressure (will also have splenomegaly due to pooling of cells)
altered RBC membranes caused by abnormal blood lipid content in liver disease → spleen will cull or pit these abnormal cells
associated liver disease
alcohol intoxication
direct toxic effect of ethanol on the developing erythroblasts
how is stimulated erythropoiesis/reticulocytosis associated with nonmegaloblastic anemia?
increased EPO in the presence of an adequate iron supply can result in the release of shift reticulocytes → cells are larger than normal, increase in polychromasia, and oval macrocytes are not present
what is the pathogenesis of hereditary spherocytosis (hemolytic anemia)?
patients will have defects in the membrane cytoskeleton (spectrin, ankryin, band 3, or band 4.2) → since red cell membranes are fragile, the RBCs cannot squeeze through the spleen properly which causes the formation of spherocytosis (eaten by macrophages)
patients who have it mild do not need treatment but it if it severe, a splenectomy can be useful
what would the RBC morphology look like in those with hereditary spherocytosis?
red cells are normochromic and normocytic… depending on patient’s particular genetic defect, there could be a ton of spherocytes (smaller than RBCs and lack central pallor) or just a few
what is the pathogenesis of G6PD deficiency (hemolytic anemia)? (learning objective)
G6PD helps reduce nasty free radicals made during cell metabolism… without enough G6PD around, the free radicals attack the molecular bonds between heme and globin becomes denatured and forms heinz bodies → spleen will pit the heinz bodies, creating a bite cell
is an intrinsic defect in which the RBC enzymes for maintaining Hb and membrane sulfhydryl groups are deficient in the reduced state for maintaining adequate levels of ATP for cation exchange causing hemolytic anemia
those who have a deficiency of G6PD are normally ok until they encounter some sort of oxidizing substance like a drug or fava beans
patient must avoid exposure to known oxidants but the hemolysis is self-limiting so it will spontaneously resolve in a week or so
what would the RBC morphology look like in those with G6PD deficiency?
patients going through an acute hemolytic episode will have fragmented cells, microspherocytes, and bite cells → supravital staining will reveal the Heinz bodies
patients who have not been exposed to offending agents will have no anemia
heinz bodies will decrease in number as Hb bottoms out because younger cell have greater G6PD activity
what are hemoglobinopathies? (learning objective)
involves abnormal hemoglobin (particularly the beta-chain) and could occur in homozygous or heterozygous forms
what is sickle cell anemia? (learning objective)
an SNP in the beta chain where glutamic acid is replaced by valine at the 6th position → effects of this SNP is a chain in the net charge and electrophoretic mobility which helps ID on Hb electrophoresis
serious chronic hemolytic anemia due to extravascular hemolysis (increased removal of abnormal cells by the RE system)
is the most symptomatic hemoglobinopathy and occurs in 1 of every 365 African American births
what are the screening tests to see if a patient has HbS?
metabisulfite reduction → reducing agent and enhances deoxygenation and sickle cell formation but does not differentiate between heterozygous or homozygous
the sickle dex test → HbS is less soluble than HbA under decreased oxygen tension which in turn causes the sample to turn opaque but CANNOT tell if a patient has the disease or just the trait — just if the patient makes HbS
HbS is released from lysed RBCs and reduced by sodium dithionite
false positives can occur from protein disorders
false negatives come from only low amounts of HbS being present
what would sickle cell trait and disease look like when using electrophoresis (confirmatory test)? (learning objective)
normal
will have A2 (<3.7%), HbF (<2%), and HbA
sickle cell trait
will have A2, HbS (35-45%), HbF, and HbA (50-65%)
sickle cell disease
will have A2, HbS, HbF, no HbA
remember, a control must be used in electrophoresis too
what are the various treatments in sickle cell anemia? (learning objective)
avoid dehydration and infection which both cause vaso-occlusion resulting in sickling (preventative)
blood transfusion (in aplastic crisis or splenic sequestration)
could be helpful in preventing complications of SS anemia by suppressing the formation of new HbS by the BM
hydroxyurea → drug that reduces intracellular sickling
BM transplant → potential cure
what is the clinical manifestation of sickle cell anemia? (learning objective)
hemolysis and severe pain in the abdomen, bone and joints due to the occlusion of small blood vessels by sickle cell infarcts, otherwise known as a sickle cell crisis
even at physiologic oxygen tension, sickle cell may form
what is the pathogenesis of sickle-cell anemia (hemoglobinopathies)? (learning objective)
abnormal hemoglobin (HbS) in sickle cell anemia will change shape when it releases oxygen, causing it to polymerize and in turn distorting it into a sickle shape
sickle cells are fragile and will stick together in small vessels (capillaries), leading to ischemia & tissue hypoxia because of a blockage → cells are phagocytized by macrophages (causes the anemia) and the tiny, repeated infarcts in the spleen leads to fibrosis and eventual autosplenectomy
prevent triggers like things that will make RBCs want to give up O2 and vaccination in those who have undergone autosplenectomy
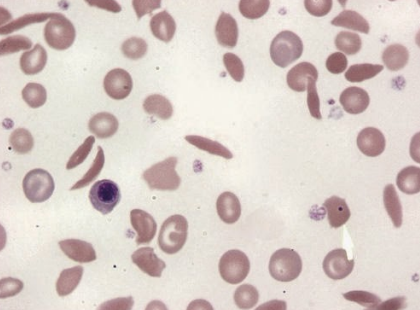
what would the RBC morphology look like in those with sickle-cell anemia? (learning objective)
will often show normocytic, normochromic cells
during times of crisis/decreased oxygenation, there will be sickled cells in the blood
when autosplenectomy occurs, there will be a post-splenectomy blood picture which includes NRBCs, polychromasia, Howell-Jolly bodies (due to removal of or a nonfunctioning spleen), sickled cells, and Pappenheimer bodies
what is sickle cell trait? (learning objective)
heterozygous meaning they are a carrier → 8-10% of american blacks are affected (most common hemoglobinopathy in the US) and patient is usually asymptomatic
clinical symptoms → could appear under conditions of hypoxia: respiratory infections, CHF, anesthesia
what is hemoglobin C? (learning objective)
substitution of lysine for glutamic acid in number 6 position of beta chain and could also be heterozygous or homozygous → mild hemolytic anemia and is mostly asymptomatic with occasional jaundice
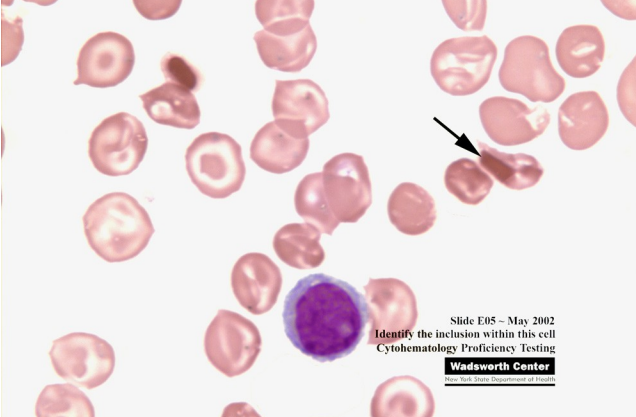
what would the RBC morphology look like in those with HbC? (learning objective)
normocytic, normochromic anemia with a high number of target cells (40-90%) and is the most striking feature of HbC… will also have the presence of intracellular rod-shaped crystals (HbC crystals)
what would the electrophoretic profile be in HbC disease and trait?
HbC disease
HbA → 0%, HbC → almost 100%, HbF → <7%
HbC trait
HbA → 60-70%, HbC → 30-40%
what is hemoglobin C trait? (learning objective)
heterozygous state (many times they aren’t anemia) → asymptomatic with target cells up to 40% but no HbC crystals
what are heterozygous hemoglobinopathies? (learning objective)
inheritance of different abnormal hemoglobins from each parent results in interactions producing hemolytic anemia of varying disease → HB SC disease which is sickle cell - thalassemia disease (Hb S/beta-thalassemia)
frequency is similar to sickle cell disease and the severity of the clinical manifestations is intermediate between sickle cell disease and trait
sickle cell prep is positive
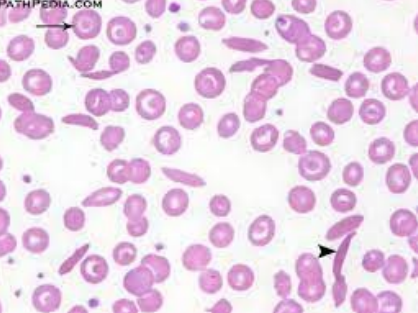
what would the RBC morphology look like in those with Hb SC disease? (learning objective)
anemia will vary from mild to moderate with normocytic, normochromic RBCs, numerous target cells (<85%) and odd shaped cells… will look like there may be sickle cells (rare) and HbC crystals
what is the electrophoretic profile in HB SC disease? (learning objective)
HbA → 0%, HbC and HbS → equivalent, HbF → up to 7%
what is the situation with sickle cell - thalassemia disease (HbS/beta-thalassemia)? (learning objective)
situations of complete absence of beta-chain production (B0) are more severe than marked decrease (B+)… will have positive sickle cell prep and solubility tests
what is the electrophoretic profile in those with S/B0 thalassemia? (learning objective)
HbA → 0%, HbS → 75-90%, HbF → 5-20%, HbA2 → >4.5%
what is the electrophoretic profile in those with S/B+ thalassemia? (learning objective)
HbA → 15-30%, HbS → >50%, HbF → 1-20%, HbA2 → >4.5%
what is the pathogenesis of thalassemia? (learning objective)
quantitative disease of Hb (hereditary usually occurring in Mediterranean ancestry like Greece) → impaired production of one or both of a pair of polypeptide chains found in Hb molecules
structure is normal but there is a decreased rate of synthesis which results in less than normal number of chains and fewer complete Hb molecules
in thalassemia, patient ends up with a two-fold problem
decreased hemoglobin production (due to decrease in globin chains)
excess unpaired alpha chains (in beta) or beta, gamma, and delta chains (in alpha) which form tetramers and lead to premature RBC destruction
mild thalassemia doesn’t require treatment but those with severe anemia, patients may need repeated RBC transfusions or even BM transplantation
what is the pathophysiology of beta thalassemia? (learning objective)
beta-chain synthesis is impaired causing an excess of alpha chains which precipitate to damage the RBC membrane → spleen will cull them
precipitate filled RBCs could be destroyed in the BM by macrophages which results in a large degree of ineffective erythropoiesis
circulating RBCs with precipitates are removed by the spleen → causes chronic hemolysis
what would the RBC morphology look like in those with thalassemia? (learning objective)
microcytic, hypochromic anemia with a decrease in MCV, MCH, and usually MCHC and an increase in RDW (>14.5%) → will have target cells and microcytosis, basophilic stippling and NRBC (meaning there is aniso and poik)
reticulocytes (could be stressed retics due to the anemia) and bilirubin usually increased (higher destruction of RBCs)
RBC count is normal or increased for the degree of anemia but will have a decrease in hemoglobin
what is alpha-thalassemia major (also called hemoglobin Barts or hydrops fetalis)? (learning objective)
complete absence of alpha chains which is incompatible with life and is homozygous, meaning both parents must at least carry alpha-globin gene deletions
no hemoglobin is produced due to alpha chains being required for the formation of hemoglobin
infants are still born/die in utero due to the child being unable to make normal hemoglobin to carry oxygen throughout the body
all four genes for alpha are missing
what counts as hemoglobin Bart’s?
Hb that lacks alpha chains → at least one gene missing
what is alpha-thalassemia-hemoglobin H disease and how would it be treated? (learning objective)
symptomatic (similar to anemia and chronic hemolysis) but nonfatal (normal life expectancy) and occurs when 3 of 4 alpha-genes are deleted → splenomegaly will be present
treatment would be long-term transfusion therapy and splenectomy (alleviates the anemia when it is removed)
what would the RBC morphology look like in those with alpha-thalassemia-hemoglobin H disease? (learning objective)
microcytic, hypochromic anemia with Hb 8-10 g/dL and will have increased reticulocytes and NRBCs will be present (5-10%)
what is alpha-thalassemia-minor (trait) and what is the treatment? (learning objective)
occurs when 2 of 4 alpha-genes are missing and is found in all geographic locations… patients are asymptomatic with mild anemia
there is a measurable decrease in the production of alpha-containing hemoglobin BUT unaffected globin genes are able to direct synthesis of globin chains faster than normal and compensate for the affected genes
patients do not require medical intervention and has a normal life span
what is the genetics of thalassemia? (learning objective)
mom is a silent carrier and dad has thalassemia trait
a silent carrier → only one gene is missing
thalassemia trait → two genes are missing
HbH disease → three genes missing
no anemia → no genes missing
child wins the genetic lottery
being a silent carrier or having trait or disease = Hb Barts
what would the what would the RBC morphology look like in those with alpha-thalassemia-minor? (learning objective)
microcytic, hypochromic anemia with a few target cells but could be masked by iron deficiency anemia… to tell the difference, the persistence of microcytes following treatment of iron deficiency anemia is suggestive of thalassemia
what is beta-thalassemia major (homozygous) or Cooley’s anemia? (learning objective)
extreme form of thalassemia where beta-chains cannot be made and is associated with physical abnormalities… onset is generally in infancy with symptoms of irritability, pallor (blue characteristics), and failure to thrive
anemia will place a tremendous burden on the cardiovascular system due to the increased pumping of blood to compensate for tissue hypoxia… if undetected, cardiac failure can occur
physical abnormalities → growth is slowed, brown pigmentation of skin, chronic hemolysis which often produces gallstones, gout, and icterus (due to bilirubin build up)
will have facial/skull deformities as well due to the marrow cavities enlarging in every bone to accompany the hyperplastic marrow
will have extramedullary hematopoiesis (spleen, liver) to make more RBCs to compensate… spleen may become enlarged and congested with abnormal RBC
what are the other symptoms of homozygous beta-thalassemia?
increased metabolic rate
fever
lethargy
weakened musculature
decreased body fat
decreased appetite
infections (common cause of death)
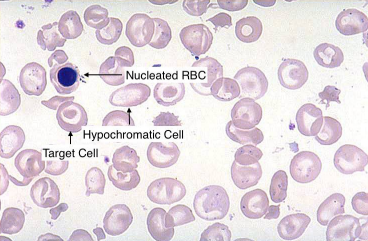
what would the what would the RBC morphology look like in those with homozygous beta-thalassemia? (learning objective)
HB LEVEL IS LOW WITH 2-3 g/dL → anemia is markedly microcytic and hypochromic (MCV <67 with markedly reduced MCH and MCHC and RDW may be increased)… marked aniso and poik and retics are not as increased as they should be because of ineffective erythropoiesis and NRBCs will be present
secondary leukopenia (decreased leukocytes) and thrombocytopenia (decreased thrombocytes) → will become trapped in the enlarged spleen and is a secondary effect of the spleen
increased unconjugated bilirubin (will not show up in urine)
body is adapted to the low amounts of Hb and Hb electrophoresis is helpful in diagnosis
what is the treatment for homozygous beta-thalassemia? (learning objective)
regular transfusion program
prolongs in life in the second or third decade but reduces developmental and growth delays
iron chelating agents are given to decrease deposition of iron in the tissues
splenectomy
for patients over 5 → makes them susceptible to infections
prognosis
untreated patients will die in the first or second decade of life
patients in hypertransfusion therapy can help extend their life by at least a decade
second decade patients will develop endocrine disorders (diabetes), and hepatic and cardiac disturbances from excessive iron deposits
what is heterozygous beta-thalassemia minor and the treatment? (learning objective)
heterozygous inheritance meaning they can still produce beta-chains → normal beta-gene directs synthesis of sufficient amounts of beta-chains to make enough HbA for near normal oxygen delivery and RBC survival
patients appear to be asymptomatic except during periods of stress like pregnancy, infection, and folic acid deficiency
can develop a moderate microcytic anemia under these conditions
patients generally do not require treatment due to being asymptomatic (mostly) and will have a normal life expectancy
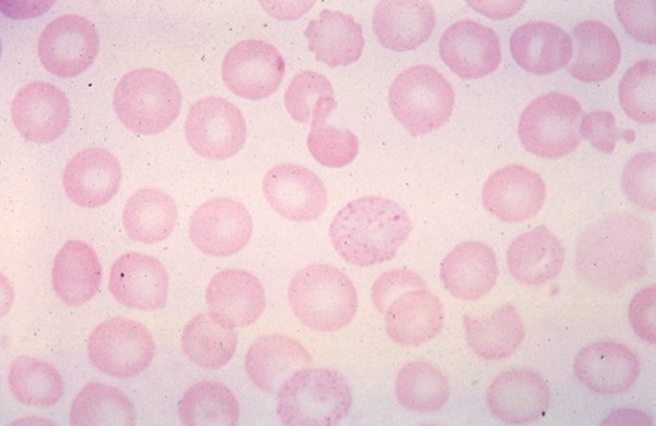
what would the what would the RBC morphology look like in those with heterozygous beta-thalassemia? (learning objective)
Hb is 9-14 g/dL which is reduced but not as significant → microcytic, hypochromic RBCs as in IDA with low MCV and MCH… MCHC may be low and RDW is normal, RBC count is elevated but values for Hb and HCT are reduced
mild to moderate degree of poik, target cells, and basophilic stippling are often found
what are hemolytic anemias? (learning objective)
a group of normocytic, normochromic anemias in which RBC is prematurely destroyed and can be classified according to various things:
source of the defect
intrinsic (immune complex) or extrinsic
mode of onset
inherited or acquired
location of hemolysis
intra- (occurring in the vessels and would appear in the urine) or extra- (spleen, liver won’t see in urine)
type of poikilocyte
schistocytes or spherocytes
what is a constant feature of hemolytic anemia?
reticulocytosis → compensated hemolytic disease in which the BM is able to increase erythropoiesis to compensate for the decreased RBC life span (RBC does not develop)
will develop into anemia when → RBC destruction accelerates beyond the compensatory capacity of the marrow (hemolytic crisis) and when the marrow suddenly stops producing RBCs (aplastic anemia)
what is the clinical findings in hemolytic anemias? (learning objective)
pallor, fatigue, and cardiac symptoms
jaundice due to increase in bilirubin production
gallstones due to bilirubin
dark or red urine due to the excretion of plasma hemoglobin (intravascular hemolysis)
splenic hypertrophy → extravascular hemolysis
what is intravascular hemolysis? (learning objective)
RBC is destroyed within the blood vessels
Hb is bound to the transport protein → haptoglobin
too big to pass through the glomerulus
when haptoglobin is depleted, hemopexin complexes with Hb
too big to pass through the glomerulus
when haptoglobin and hemopexin are depleted, free Hb appears in the urine (pink, red or brownish black) due to exceeding the renal threshold
filtered Hb may be reabsorbed in the PCT
what must happen in order for an RBC to go through intravascular hemolysis and what may it be caused by? (learning objective)
to undergo intravascular hemolysis (happens in the vasculature), RBCs must be severely damaged and may be caused by:
activation of complement on the RBC membrane (pierces the membrane)
physical or mechanical trauma to the RBC
presence of soluble toxic substances in the RBCs environment
what is the lab findings in intravascular hemolysis? (learning objective)
hemoglobinuria
hemoglobinemia
decreased haptoglobin
decreased hemopexin
increased LDH-enzyme → present in high concentration in RBCs
what is extravascular hemolysis? (learning objective)
RBCs are removed from the circulation by phagocytes in the tissue and more common than intravascular hemolysis (main mode of hemolysis and Hb will be in macrophages) → may occur in the phagocytes of the spleen, liver, or bone marrow
no hemoglobinemia and hemoglobinuria
Hb is degraded with the phagocyte to heme and globin
heme is catabolized to iron, biliverdin and CO2 → biliverdin to bilirubin (plasma), binds to albumin, and is excreted by the liver (ex. kernicterus)
BM macrophages are responsible for the removal of maturing precursor cells that are intrinsically abnormal → cytoplasmic or nuclear maturation abnormalities due to the high degree of ineffective erythropoiesis (iron deficiency and megaloblastic anemia)
what does the type and degree of RBC damage determine in extravascular hemolysis?
determines the primary site of erythrocyte destruction
spleen is more efficient at removing slightly damaged RBCs
liver is an important site for the removal and phagocytosis of extensively damaged RBCs
whats a common cause of hemolytic anemia associated with extravascular hemolysis? (learning objective)
antibodies directed against the RBC → antibody and complement attached to the cell membrane making it a target for removal from the circulation by phagocytes
what does the site and extend of extravascular hemolysis dependent on?
class of antibody and the presence of absence of complement
complement and IgM → removed by the liver due to C3b receptor
driver for transfusion reactions
IgG → removed by macrophages of the spleen
what are the lab findings in hemolytic anemias? (learning objective)
increase in expired CO
increased RBC destruction results in increase heme catabolism and increased production of CO
increase in carboxyhemoglobin (COHb)
COHb is a stable complex of CO that forms in RBCs when CO is inhaled
increase in bilirubin and urobilinogen
haptoglobin and hemopexin levels may also be decreased → in severe or chronic conditons
what are intrinsic defects in hemolytic anemias? (learning objective)
abnormality of the RBC itself → most are hereditary and is in extravascular hemolysis… cells are more susceptible to damage by environmental factors
structural defects may cause the membrane to become abnormally permeable, rigid, or unstable and easily fragmented and ultimately hemolysis
what is paroxysmal nocturnal hemoglobinuria? (learning objective)
an intrinsic defect in which the RBC lacks a protein that controls complement attachment and activation (acquired abnormality)
will have normocytic or macrocytic: microcytic, hypochromic if Fe deficient
what are extrinsic defects in hemolytic anemias? (learning objective)
usually acquired → RBCs are damaged by chemical, mechanical, or physical agents and hemolysis could be intravascular or extravascular
what are chemical extrinsic effects in hemolytic anemias? (learning objective)
substances in the circulation that are toxic to the cell and causes direct cell hemolysis or alter cell membranes and lead to removal of the cell in the spleen
what are mechanical trauma extrinsic effects in hemolytic anemias? (learning objective)
trauma to the RBC may cause the RBC to fragment which produces striking abnormalities on the blood smear… splenomegaly may cause anemia to hyper-sequestration of RBC
what are physical agent extrinsic effects in hemolytic anemias? (learning objective)
antibodies or complement attached to the RBC membrane sensitized the cell which caused premature removal by macrophages in the spleen or liver… can be autoimmune, alloimmune, or drug induced
what do all immune anemias have? (learning objective)
all have a positive DAT due to immunoglobulin or complement on the cell
what is the pathogenesis of autoimmune hemolytic anemia? (learning objective)
warm
patient makes IgG anti-RBC antibodies which binds at 37C best which is warm
splenic macrophages will think that the antibody-coated RBCs are appetizing and will either cull or pit them
cold
patient makes IgM anti-RBC antibodies that bind at <37C which means they will bind in distal body parts but fall off in warm body parts
IgM will bind a bunch of RBCs together which forms clumps and complement will bind to the RBCs too → causes intravascular hemolysis