MCB topic 5.2
1/53
Earn XP
Description and Tags
Weeks 10-11
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
54 Terms
Cardinal signs of inflammation
Heat (calor)
Redness (Rubor)
Swelling (tumor)
Pain (Dolar)
Loss of Function (Functio laesa)
Purpose of inflammation?
Inflammation is a protective response intended to eliminate the cause and consequence (necrotic cells and tissue) of cell injury.
Dilute
Destroy
Neutralise
Initiate resolution (repair)
Inflammation is fundamental to disease
Process that destroys and eliminates microbes and necrotic tissue but can also injure normal tissue.
Without inflammation infections would go unchecked and wounds would not heal.
Examples of ‘-itis’

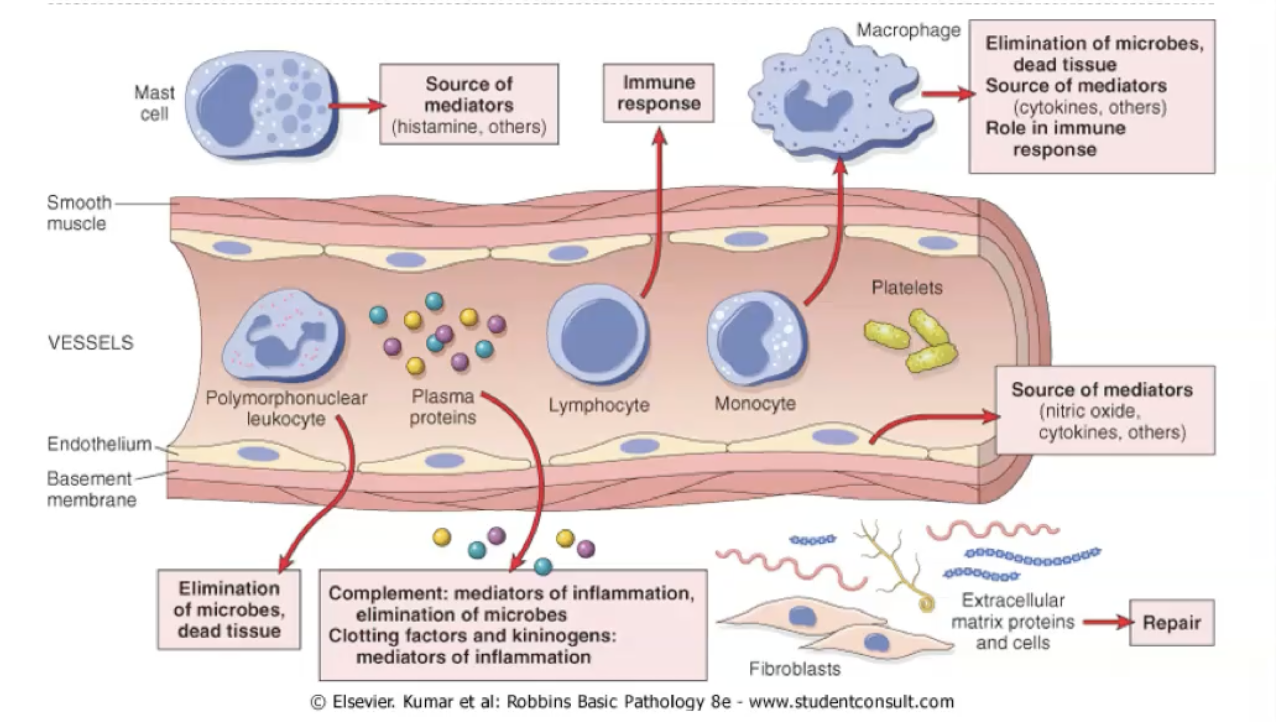
What are the components of inflammation?
Acute vs chronic inflammation (quick comparison)?
ACUTE:
Rapid onset, short duration.
Fluid, plasma protein and cellular exudate.
Neutrophilic leukocyte accumulation.
CHRONIC:
Insidious onset.
Long duration.
Lymphocytes and macrophages.
Scarring.
Acute inflammation purpose?
Acute inflammation: recognition, recruitment, removal, resolution, regulation.
Acute inflammation is required for wound healing.
Acute inflammation can eliminate the consequence of injury, including dead cells.
Acute inflammation can injure normal tissue.
Acute inflammation can eliminate the cause of injury such as microbes.
Compared to acute inflammation, chronic inflammation has a longer duration and is associated with the presence of macrophages and lymphocytes.
Acute inflammation: recognition
Recognition: a protective response intended to eliminate the cause and consequence of cell injury. Host must recognise the cause (infections, foreign bodies, immune reaction) or consequence (necrotic cells and tissue) of injury - we know the consequences of injury can be dead cells.
Phagocytes and dendritic cells (cells that reside in the connective tissue of organs) and many other cells (epithelial cells) express receptors that sense the presence of microbial pathogens and substances released from dead cells.
Pattern recognition receptors (PRR): toll like receptors: recognise patterns that are unique to bacteria, viruses, and other pathogens.
Pathogen-associated molecular patterns (PAMPs): toll-like receptors - recognise patterns that are unique to bacteria, viruses, and other pathogens - e.g., lipopolysaccharide (LPS), Lipoteichoic acid, unique surface glycans, and viral RNA.
Damage-associated molecular patterns (DAMPs): inflammasome - recognises products of dead cells and some microbial products - e.g., uric acid (DNA breakdown), ATP (mitochondria), and decrease in potassium ion concentrations (ion channels).
Pattern recognition receptors detect the cause or consequence of injury and release of chemical mediators that initiate vascular and cellular changes that lead to the recruitment of leukocytes to the site of injury. They would be expected to find a pattern recognition receptor that would detect an extracellular bacteria on a macrophage, epithelial cells, dendritic cells, and endosomes.
Acute inflammation: recruitment
Vascular changes: vasodilation, vascular permeability, stasis. Rapid response designed to deliver leukocytes and plasma protein to the site of injury.
Cellular: margination, rolling, adhesion, transmigration, migration/chemotaxis.
Normal hydrostatic pressure: fluid flow
From areas of high hydrostatic pressure (HP)
Leaves the arterial end of the capillary network (32mmHg).
Reabsorbed at the venous end (12mmHg).
Towards areas of high osmotic pressure.
Net flow is negligible across the vascular bed.
Transient vasoconstriction
Arteriolar vasodilation
Chemical mediators (histamine and NO).
Increased blood flow.
Engorgement of capillary beds.
Erythema (redness) and warmth.
Transudate
Increased hydrostatic pressure.
Accumulation of interstitial fluid.
Ultrafiltate.
Low protein concentration, few cells.
Vascular permeability
Endothelial cells line the entire blood and lymphatic vascular system, from the heart to the smallest capillary, and control the passage of materials - and the transit of WBC - into and out of the bloodstream.
Vasoactive mediators:
Immediate and transient: histamine, bradykinin, leukotrienes, and other chemical mediators. Endothelial cell contraction, short lived (minutes).
Slow, prolonged: IL-I and TNF. Changes in cytoskeleton. Prolonged (hours to days).
Endothelial injury:
Direct injury of endothelial cells.
Delayed: mild injury, e.g., sunburn.
Immediate: severe injury, e.g., burn or infection of endothelial cells.
Sustained: hours to days (until injury is repaired).
Exudate:
Increased vascular permeability.
Exudate (protein rich fluid)
Change in osmotic pressure.
Outflow of water and ions to extravascular space.
Edema-fluid accumulation in extravascular space.
Vascular change
Transient vasoconstriction.
Arteriolar vasodilation
Chemical mediators (histamine and NO)
Increased blood flow.
Engorgement of capillary beds.
Erythema (redness) and warmth.
Transudate (swelling)
Increased vascular permeability
Chemical mediators (histamine, bradykinin, leukotrienes, or direct injury)
Contraction or injury of endothelial cells.
Exudate (swelling)
Relation of cardinal/classic signs of inflammation to changes
Heat
Vasodilation; increased blood flow to the injured region.
Redness (erythema)
Vasodilation; congestion / hyperaemia / engorgement
Swelling
Vasodilation and vascular permeability leading to extravasation of fluid (transudate / exudate / edema)
Pain
Compression of tissues.
Direct effect of inflammatory mediators.
Loss of function
Direct effect of injury
Pain / swelling.
Vascular change - stasis
Dilated small vessels and increased vascular permeability.
Extravasation of fluid (transudate and exudate)
Increased RBC concentration.
Slow flow
Stasis is the abnormal slowing or stagnation of blood flow, usually caused by vasodilation, venous obstruction, immobility, or increased blood viscosity, and it leads to concentrated red cells, sluggish movement of plasma, and the drifting of leukocytes toward the vessel wall. This slowdown is crucial in acute inflammation because it promotes leukocyte margination, allowing white cells to roll, adhere, and exit the bloodstream, and it also contributes to thrombosis by allowing clotting factors and platelets to accumulate. Clinically, stasis underlies conditions like venous congestion in heart failure, deep vein thrombosis during immobility, and chronic venous insufficiency, all of which reflect impaired forward flow and reduced tissue oxygen delivery.
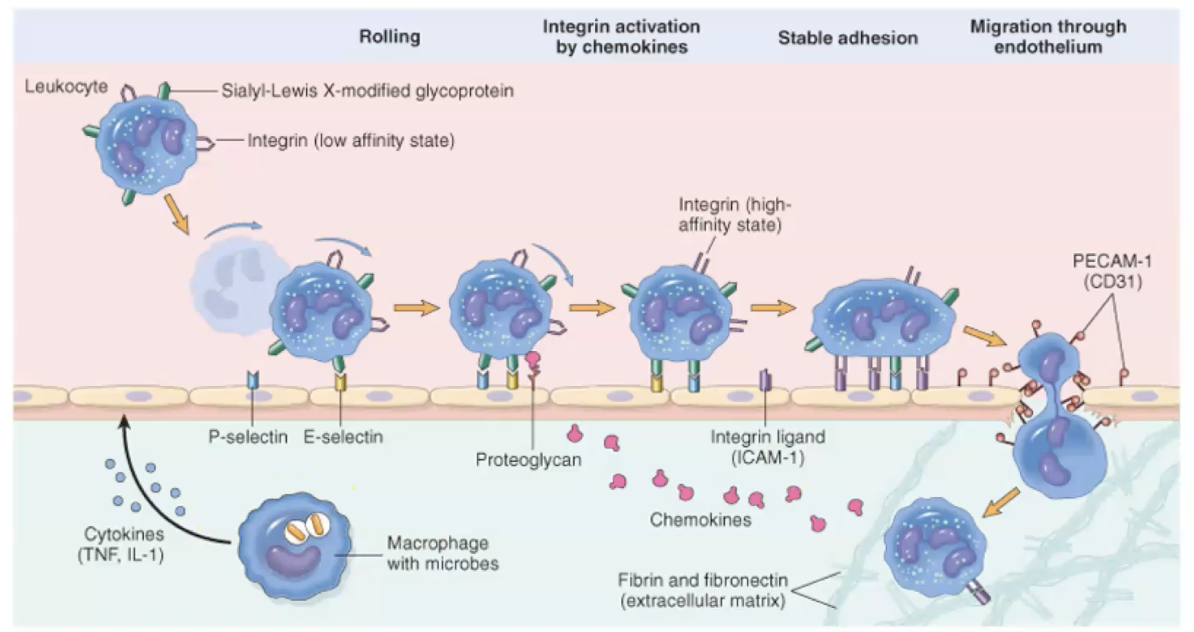
Recruitment - cellular changes (there are five steps)
Margination:
Slowed blood flow-stasis.
Leukocytes are pushed to the 'margins' of the blood vessels.
Tumble along the endothelial surface - 'rolling'
Rolling
Weak transient adhesion.
Reduces rolling velocity.
Mediated by selectins (bind sugars)
Sialylated oligosaccharides expressed on glycoproteins.
Low expression levels or absent normal endothelium.
Upregulated by chemical mediators in response to infection or tissue injury.
Adhesion
Mediated by integrins
Expressed on leukocyte plasma membrane.
Low affinity until activated by chemokines.
Leukocyte activation -> clustering of integrins -> high affinity.
Inflammatory cytokines stimulate endothelial cell expression of integrin ligands (I-CAM, V-CAM)
Stable attachement of leukocytes to endothelial cells at the site of inflammation.
Transmigration
Transmigration of diapedesis-movement of leukocytes between cells at the intercellular junctions.
In response to chemical gradient produced at the site of inflammation.
Postcapillary venule.
Migration / chemotaxis
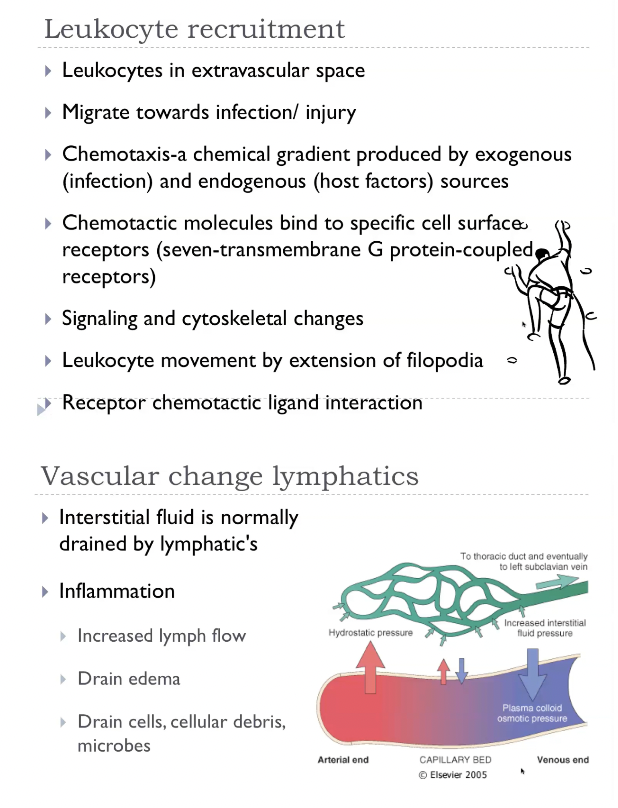
AI summary of the cellular changes for recruitment
During inflammation, a white blood cell leaves the bloodstream and enters the tissue in five steps:
Margination —
Blood flow slows down, so white blood cells drift from the centre of the vessel toward the edges, closer to the vessel wall.Rolling —
The white blood cell sticks and lets go, sticks and lets go, like Velcro touching lightly. This makes it “roll” along the inside of the vessel.Firm adhesion —
Chemical signals activate the white blood cell, making its “sticky proteins” stronger. Now it sticks firmly to the vessel wall and stops moving. IntegrinDiapedesis / transmigration —
The white blood cell squeezes between the endothelial cells, slipping out of the blood vessel like squeezing through a narrow doorway.Chemotaxis —
Once outside, it follows chemical signals (like a scent trail) to move toward the exact site of infection or injury.
Transudate vs exudate AND selectins vs integrins
TRANSUDATE: a low protein, ultrafiltrate that accumulates in the interstitial tissue in response to vasodilation and increased hydrostatic pressure.
EXUDATE: a protein rich fluid that accumulates in the interstitial tissue in response to increased vascular permeability and changes in osmotic pressure
SELECTINS: molecules expressed on the surface of leukocytes and endothelial cells that reduce the velocity of leukocytes during the recruitment stage of acute inflammation. P and E selectins.
INTEGRINS: molecules expressed on the surface of leukocytes and endothelial cells that reduce the velocity of leukocytes during the recruitment stage of acute inflammation. Stronger affinity.
Acute inflammation - removal
Phagocytes express receptors that sense the presence of microbial pathogens and substances released from dead cells.
Toll-like receptors - recognise patterns that are unique to bacteria, viruses, and other pathogens.
NOD-like receptor 3, NLRP3-inflammasome - recognises products of dead cells and some microbial products.
Recognition and subsequent phagocytosis is enhanced by opsonisation. The target particles are opsonised.
Bacterial LPS activates complement, generating C3b with opsonising properties. Antibodies bound to antigen activate complement and also act as opsonins. Collectins which bind microbial cell-wall sugar groups.
LEUKOCYTE ACTIVATION: Engagement of the receptors expressed on leukocyte surfaces by microbial products and mediators of inflammation.
Activation enhances a number of cellular functions, including:
Phagocytosis.
Intracellular destruction of phagocytosed microbes and debris.
Release of substances that destroy extracellular microbes and degrade tissue.
Produce cellular mediators to amplify the inflammatory response.
Phagocytosis and phagolysosome
Phagocytosis
Binding of opsonised particles triggers engulfment.
Receptor-initiated signalling, membrane remodelling, and cytoskeletal changes.
Actin assembly to form phagosome.
Phagolysosome
Phagosome fuses with the membrane of a lysosome.
Exposes the ingested particle to destructive process: free radicals, enzymes.
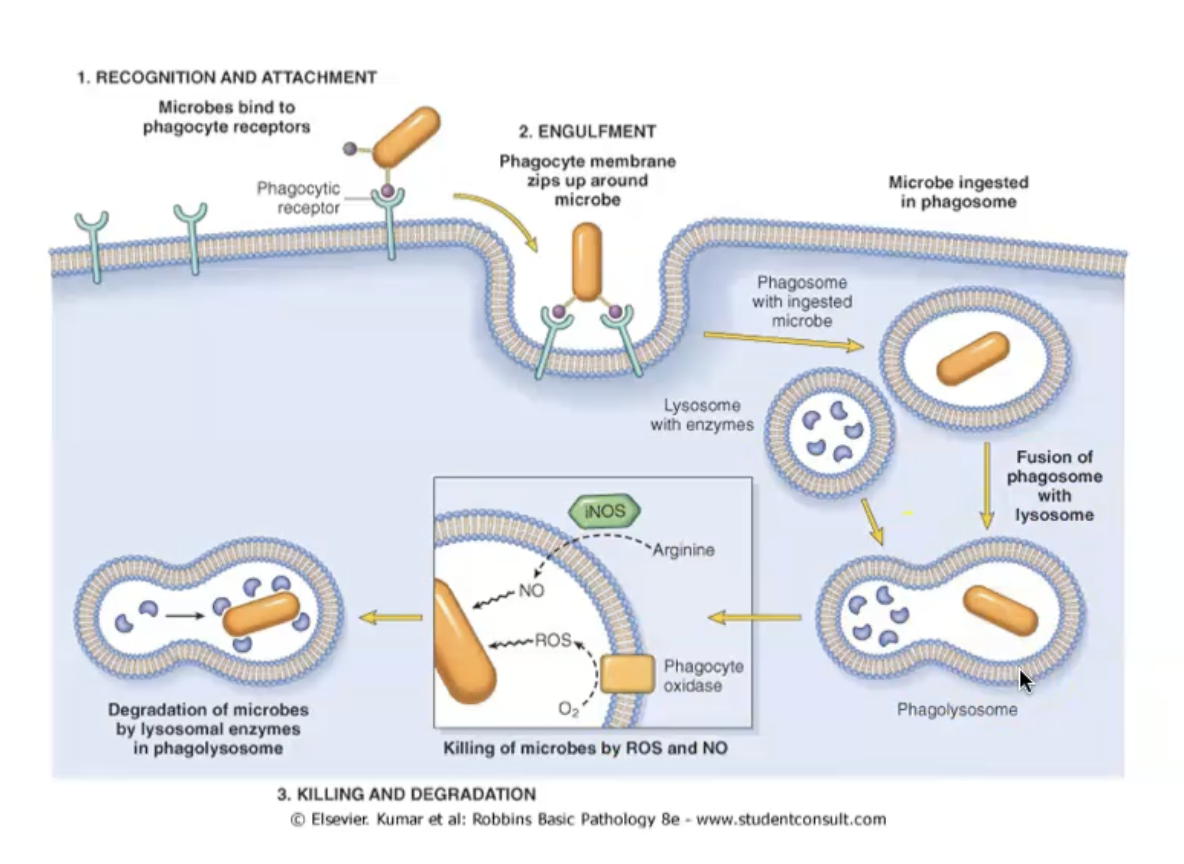
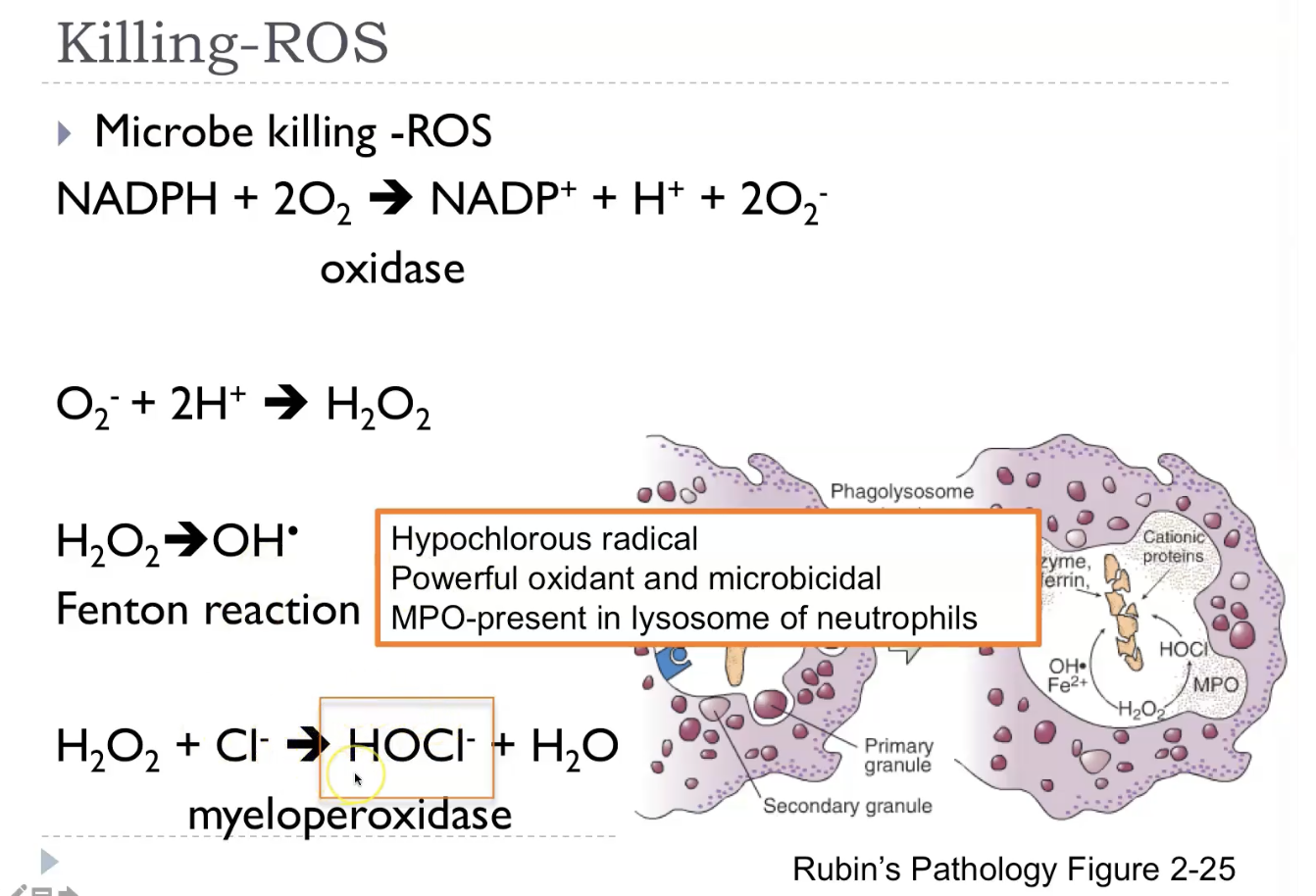
Killing - reactive oxygen species
Lysosome and phagosome fuse to form the phagolysosome.
Respiratory burst -> Increased ROS: phagocyte oxidase / NADPH oxidase.
Reactive oxygen species (ROS) are highly reactive oxygen‑containing molecules—like superoxide, hydrogen peroxide, and hydroxyl radicals—that are produced naturally during normal metabolism, especially in mitochondria, and also generated by immune cells to kill microbes. In small, controlled amounts, ROS act as signalling molecules, but when produced in excess or when antioxidant defences (like glutathione, catalase, and superoxide dismutase) are overwhelmed, they cause oxidative stress, damaging lipids, proteins, and DNA. This damage contributes to inflammation, aging, cell injury, and diseases such as atherosclerosis and neurodegeneration, while also playing a key role in microbial killing during the respiratory burst in neutrophils.
Killing - reactive nitrogen species
Reactive nitrogen species (RNS) are highly reactive nitrogen‑containing molecules—mainly nitric oxide (NO), nitrogen dioxide (NO₂), and peroxynitrite (ONOO⁻)—that are produced in cells during inflammation, especially by macrophages and neutrophils. Like ROS, they can be helpful in controlled amounts, because NO acts as a signalling molecule (regulating blood vessel dilation) and contributes to microbial killing when combined with superoxide to form peroxynitrite. But in excess, RNS cause nitrosative stress, damaging proteins, lipids, and DNA through nitration and nitrosylation reactions, contributing to inflammation, cell injury, and diseases such as neurodegeneration and chronic inflammation. They are essentially the nitrogen‑based counterparts to ROS, with similar benefits and risks.

Killing - enzyme mediated
Enzyme‑mediated reactive nitrogen species (RNS) refers to the RNS that are produced directly by enzymes, mainly nitric oxide synthases (NOS), which convert arginine into nitric oxide (NO). Once NO is made, it can react with superoxide (from NADPH oxidase) to form peroxynitrite (ONOO⁻), a powerful oxidant that damages lipids, proteins, and DNA. These enzyme‑generated RNS are used by immune cells—especially macrophages and neutrophils—to kill microbes, but in excess they cause nitrosative stress, contributing to inflammation and tissue injury. So in simple terms: enzymes make NO, and NO combines with other radicals to form harmful RNS when not controlled.

Degradation
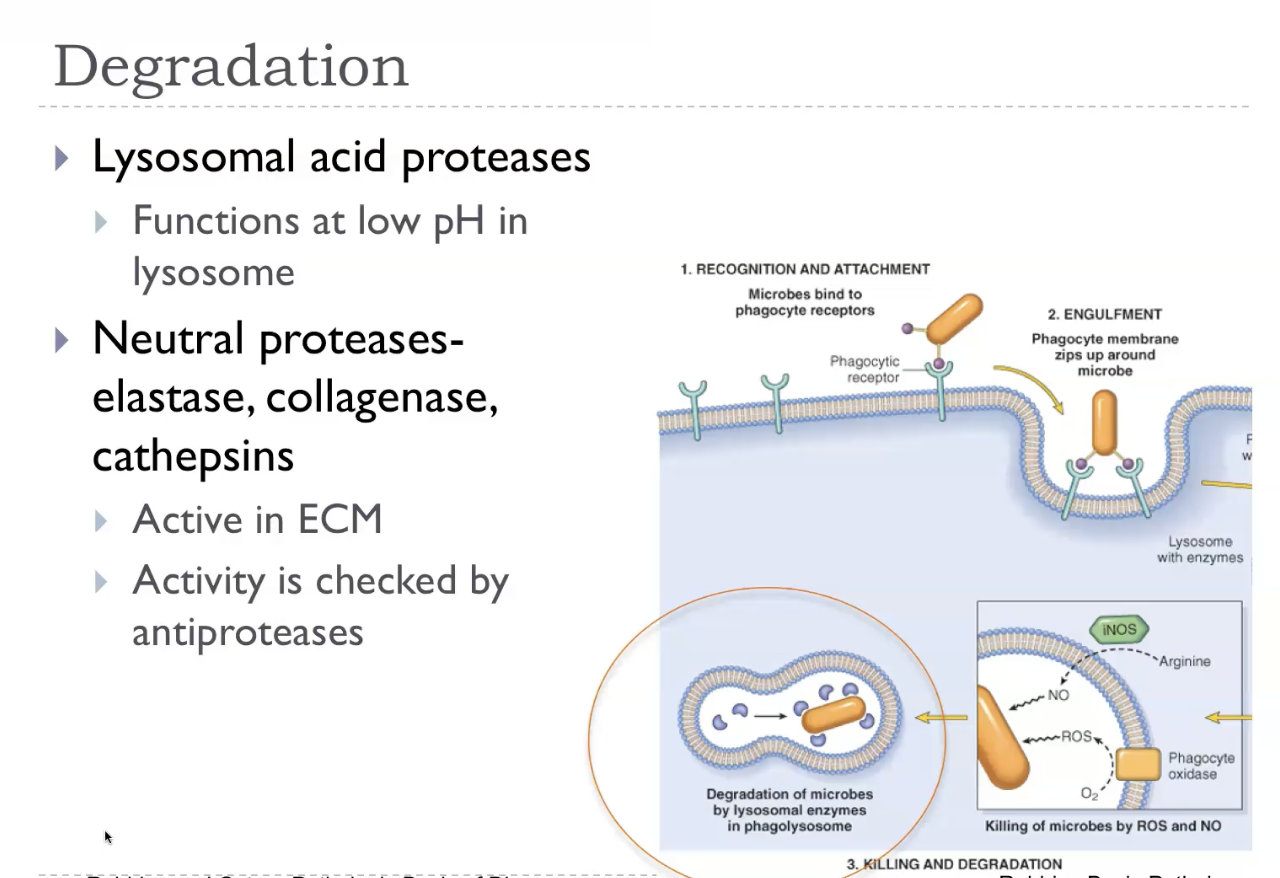
Leukocyte mediated tissue injury
Leukocyte‑mediated tissue injury occurs when activated neutrophils and macrophages release their toxic killing mechanisms—such as reactive oxygen species, reactive nitrogen species, proteases, and lysosomal enzymes—into the surrounding tissue instead of just into phagosomes. This “collateral damage” happens when leukocytes are overactivated, when the stimulus is persistent, or when they cannot fully ingest the target (like immune complexes or large parasites). As a result, the same mechanisms used to kill microbes end up damaging host cells, causing inflammation, necrosis, and worsening disease. This process underlies many conditions, including abscess formation, autoimmune diseases, asthma, and chronic inflammatory disorders.

Acute inflammation - resolution
Injury short lived. Minimal damage. Regeneration and repair of damaged tissue. Removal of dead cells. Regain function.
Neutralisation, decay, or enzymatic degradation of chemical mediators.
Normalisation of vascular permeability.
Cessation of leukocyte emigration.
Death of leukocytes (apoptosis).
Clearance of exudate (lymphatic drainage, macrophages ingestion).
Resolution is the ideal outcome of acute inflammation — it means the tissue returns completely to normal with no scarring. It happens when the injury is mild and short‑lived, and the tissue can regenerate. Resolution occurs through a coordinated process where inflammatory mediators are switched off, neutrophils undergo apoptosis and are cleared by macrophages, oedema fluid is drained via lymphatics, and the damaged cells are replaced by normal parenchymal cells. Macrophages play the key role: they clean up debris, release anti‑inflammatory cytokines like IL‑10 and TGF‑β, and restore tissue homeostasis. The end result is that the tissue looks and functions as if inflammation never happened — the opposite of scarring or chronic inflammation.
Inhibitors of inflammation
Lipoxins-inhibitors of inflammation.
AA metabolites produced by leukocytes when they enter the tissue.
Inhibit neutrophil; chemotaxis and adhesion.
Negative regulators of leukotrienes.
Inflammation is actively shut down by a set of anti‑inflammatory mediators that stop leukocyte recruitment, neutralise inflammatory signals, and promote healing. Key inhibitors include lipoxins (which block neutrophil recruitment), resolvins and protectins (omega‑3–derived mediators that promote resolution), and anti‑inflammatory cytokines like IL‑10 and TGF‑β released by macrophages to suppress immune cell activation. Other important brakes include neural reflexes (vagus nerve), antioxidants that limit ROS damage, and decoy receptors that soak up inflammatory cytokines. Together, these mechanisms ensure inflammation doesn’t overshoot and cause unnecessary tissue injury.
Outcomes of acute inflammation
Resolution:
Injury short lived.
Minimal damage.
Regeneration or repair of damaged tissue.
Functional normalcy.
Progression to chronic inflammation
If offending agent cannot be removed.
Fibrosis
Substantial tissue destruction and scar deposition.
Acute inflammation can end in resolution when the injury is mild and short‑lived, allowing the tissue to return completely to normal after neutrophils die off, macrophages clean up, and anti‑inflammatory mediators switch off the response. If the tissue cannot fully regenerate or the injury is more substantial, the outcome is healing by fibrosis, where collagen replaces damaged tissue, leaving a scar. If the cause of inflammation persists or the immune response fails to clear it, the process shifts into chronic inflammation, dominated by macrophages, lymphocytes, and ongoing tissue destruction. Finally, if the inflammatory reaction is intense and produces a lot of pus, the outcome is abscess formation, where a walled‑off collection of neutrophils and necrotic debris form
Acute inflammation - regulation
Chemical mediators
Cell derived.
Plasma derived.
Plasma derived mediators
Inter-related enzymatic cascades:
Complement system.
Kinin system.
Coagulation / fibrinolytic system.
Inactive precursors circulating in the plasma and activated at the site of inflammation.
Are safer than active mediators.
Act to amplify the response.
Each step produces end products that can have different activities.
Cell derived chemical mediators
Cell derived chemical mediators may be produced by:
Tissue macrophages.
Dendritic cells.
Mast cells
Endothelial cells.
Leukocytes.
Preformed or newly synthesised.
Acute inflammation is tightly regulated by several built‑in “off switches” that prevent excessive damage once the threat is controlled. As soon as the inflammatory trigger begins to fade, cells start producing anti‑inflammatory mediators like lipoxins, resolvins, IL‑10, and TGF‑β, which stop neutrophil recruitment and dampen cytokine production. Neutrophils undergo apoptosis, and macrophages clear them away while releasing signals that promote healing rather than inflammation. At the same time, pro‑inflammatory mediators have short half‑lives, so they naturally disappear unless the stimulus persists. The cholinergic anti‑inflammatory pathway (via the vagus nerve) also reduces TNF release, and antioxidants limit ROS‑mediated injury. Together, these mechanisms ensure that acute inflammation stays focused, controlled, and temporary.
Preformed cellular mediators
Histamine:
Preformed granules.
Released by a variety of stimuli: activation of complement, physical injury, binding of IgE, other chemical mediators such as cytokines.
Vascular changes: dilation and permeability.
Preformed cellular mediators are substances that inflammatory cells keep stored in granules so they can be released instantly when injury or infection occurs. The most important are histamine from mast cells, basophils, and platelets, which causes vasodilation and increased vascular permeability; serotonin from platelets, which has similar vascular effects; and lysosomal enzymes from neutrophils and macrophages, which can destroy microbes but also damage host tissue if released extracellularly. Because they are pre‑synthesised and stored, these mediators act immediately, initiating the early vascular and cellular changes of acute inflammation before newly synthesised mediators (like prostaglandins and cytokines) take over.
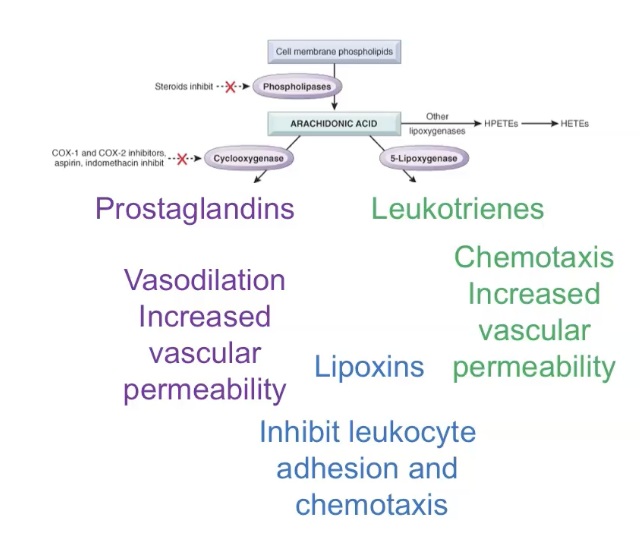
Newly synthesised mediators
Arachidonic acid metabolites - prostaglandin and leukotrienes.
AA metabolites (eicosanoids) mediate most inflammatory steps
Prostaglandins and leukotrienes.
Short range, act locally, decay spontaneously or enzymatic degradation.
Pain and fever
Mediated by AA metabolites (prostaglandin).
Aspirin and NSAIDs target AA metabolite formation to reduce pain and fever.
Newly synthesised mediators are produced by cells only after they are activated by injury, microbes, or cytokines. These include prostaglandins and leukotrienes made from arachidonic acid, which regulate vasodilation, pain, fever, and leukocyte recruitment; platelet‑activating factor, which increases vascular permeability and activates platelets; cytokines like TNF and IL‑1 that amplify inflammation and recruit more leukocytes; and chemokines that guide leukocytes to the site of injury. Because they are made fresh, these mediators shape the later and sustained phases of acute inflammation.
Acute inflammation - the systemic effects of inflammation
Acute-phase response.
Systemic release of cytokines (IL-1, IL-6, and TNF) induced by inflammatory stimuli
Microbial products.
Chemical mediators.
Clinical features of the systemic effects of inflammation
Fever (+ 1 - 4 degrees Celsius)
Rigors / chills / decreased sweating.
Myalgia (muscle pain)
Arthralgia (joint pain).
Anorexia (loss of appetite)
Somnolence / malaise (sleepy)
Increased heart rate / blood pressure.
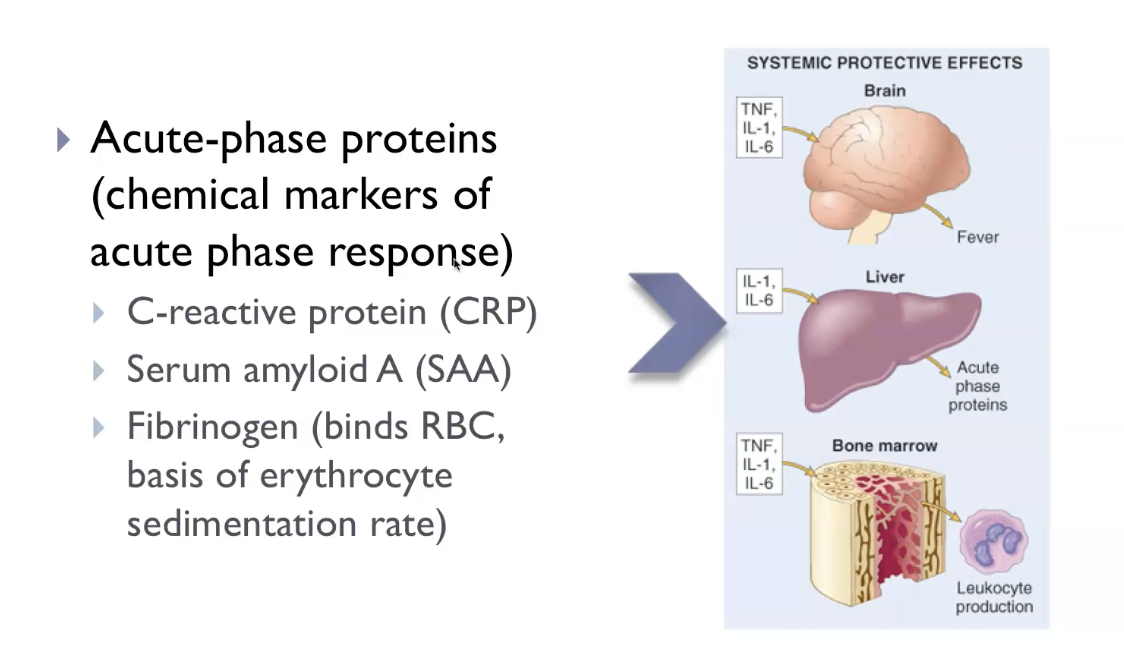
During acute inflammation, cytokines released into the bloodstream cause fever (via IL‑1 and TNF acting on the hypothalamus), increased acute‑phase proteins like CRP and fibrinogen (via IL‑6 acting on the liver), and leukocytosis, where the bone marrow releases more white blood cells to fight infection. At the same time, inflammation produces tachycardia, chills, malaise, and anorexia as part of the “sickness response.” Severe systemic inflammation can lead to septic shock, driven by high levels of TNF causing widespread vasodilation, hypotension, and metabolic disturbances. These systemic effects reflect the body’s attempt to coordinate a whole‑body defence, not just a local reaction.
Acute phase response - clinical manifestations
Fever
Elevation of body temperature.
Release of pyrogens (molecules that cause fever)
Exogenous (bacterial and viral molecules).
Endogenous pyrogens (cytokines).
Prostaglandin synthesis
Reset the temperature set point in the hypothalamus.
Thought to ward off infection and induce heat shock proteins.
Decreased sweating (redirection of blood flow to deep vascular beds).
Rigors and chills (temperature set point).
Other affects of cytokines on the brain include:
Anorexia.
Somnolence and malaise.
Muscle and joint pain.
Increased heart rate / blood pressure.
The acute‑phase response causes fever (IL‑1/TNF increase PGE₂ in the hypothalamus), malaise, anorexia, and sleepiness as part of the sickness‑behaviour pattern. The liver increases production of acute‑phase proteins such as CRP, fibrinogen, and serum amyloid A, while albumin production falls. Leukocytosis develops as the bone marrow releases more neutrophils (often with a left shift), and heart rate rises, producing tachycardia. Patients may also experience rigors/chills, sweating, and increased blood pressure due to sympathetic activation. In severe cases, excessive cytokine release can lead to septic shock, with hypotension, metabolic acidosis, and disseminated intravascular coagulation. These systemic features reflect the body’s attempt to coordinate a whole‑body defence against injury or infection.
Pathologic manifestations
*Suggests there is inflammation, but doesn’t tell us what is causing the inflammation.
The pathologic manifestations of acute inflammation arise when the normal protective response becomes too strong or persists too long, leading to tissue damage, abscess formation, ulceration, and systemic complications. Excessive neutrophil activation releases ROS, RNS, and proteases, causing collateral injury to host tissues. Intense inflammation can produce abscesses, where pus accumulates and destroys surrounding structures. Persistent inflammation on mucosal surfaces may lead to ulcers, where the epithelium and underlying tissue are eroded. Systemically, high levels of cytokines like TNF and IL‑1 can cause fever, leukocytosis, and in severe cases septic shock, with hypotension and disseminated intravascular coagulation. These pathologic outcomes reflect the destructive potential of an otherwise essential defence mechanism.
Leukocytosis vs leukopenia
Leukocytosis
Increased leukocyte count.
Neutrophilia (bacteria)
Lymphocytosis (viral infection).
Eosinophilia (hypersensitivities and parasitic infections).
Leukopenia
Decrease leukocyte counts.
Specific infections (viral / protozoan).
Overwhelming infection.
Leukocytosis is an increase in WBC count, usually caused by acute inflammation, infection, stress, or tissue injury; the bone marrow releases more neutrophils (often with a left shift), and counts commonly rise above 11,000/µL. In contrast, leukopenia is a decrease in WBC count, often due to viral infections, severe bacterial infections that exhaust marrow reserves, bone‑marrow suppression (chemotherapy, aplastic anaemia), or autoimmune destruction; counts typically fall below 4,000/µL. In simple terms: leukocytosis means the body is “gearing up” with more WBCs, while leukopenia means the body is “running low” or unable to produce enough.
Sepsis
Severe microbial infection (microorganism of their products in the blood).
Stimulates enormous production of cytokines (TNF and IL-I).
Disseminated intravascular coagulation.
Metabolic disturbances - insulin resistance and hyperglycaemia.
Hypotensive shock.
Sepsis is a life‑threatening, dysregulated systemic inflammatory response to infection, most often caused by bacteria. Massive release of cytokines—especially TNF, IL‑1, and IL‑6—leads to widespread vasodilation, increased vascular permeability, and loss of blood pressure, while activation of coagulation pathways causes microthrombi and can progress to DIC. At the same time, tissues receive less oxygen, leading to organ dysfunction in the kidneys, lungs, liver, and brain. When hypotension becomes severe and unresponsive to fluids, the condition progresses to septic shock, a medical emergency with high mortality. In simple terms: sepsis is when the body’s inflammatory response becomes so extreme that it harms the host more than the infection itself.
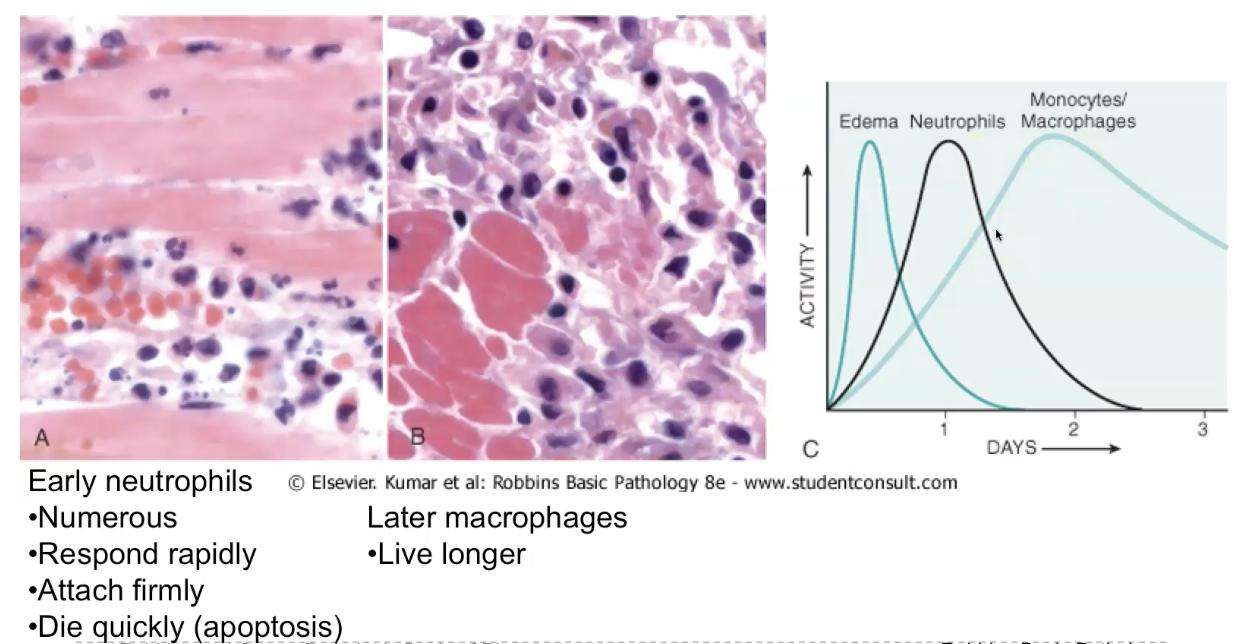
Acute inflammation - morphology
The cellular and vascular changes that characterise acute inflammation are reflected in the morphologic appearance of the reaction.
Severity.
Specific cause
Tissue type.
The morphology of acute inflammation is characterised by vascular dilation, leading to redness and warmth; increased vascular permeability, producing protein‑rich exudate and tissue oedema; and neutrophil‑rich infiltration into the affected tissue. Early on, you see congestion of dilated vessels, followed by fibrin deposition, neutrophil accumulation, and sometimes necrosis of surrounding tissue. Depending on the cause, acute inflammation may appear as serous, fibrinous, suppurative (pus‑forming), or ulcerative patterns. These morphological patterns reflect the intensity and type of injury and help pathologists identify the underlying process.
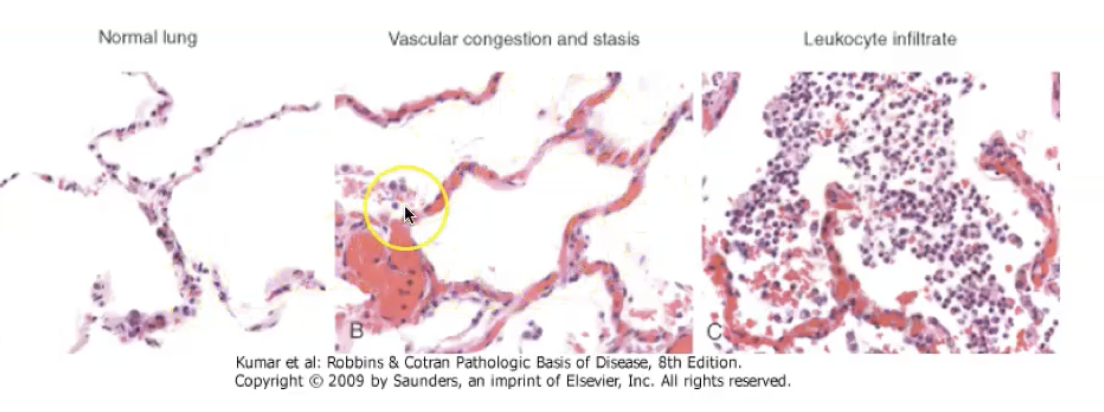
Composition of the inflammatory response
The inflammatory response is composed of vascular changes (vasodilation and increased permeability), cellular events (recruitment and activation of leukocytes—mainly neutrophils in acute inflammation), and a wide range of chemical mediators (histamine, prostaglandins, leukotrienes, cytokines, complement). Together, these components work to deliver plasma proteins and leukocytes to the site of injury, eliminate the offending agent, and initiate tissue repair. The exact composition varies with the type and duration of inflammation, but these three pillars—vessels, cells, mediators—are always present.
Morphology - serous inflammation
Watery cell poor fluid - effusion.
Derived from vasodilation:
Irritation of serous membranes.
Mesothelial cells that form the serous membrane of serous cavities.
Peritoneal (abdomen).
Pleural (lung).
Pericardial (heart).
Skin blister
Viral infection.
Burn.
Serous inflammation is characterised as a watery, cell poor effusion.
Serous inflammation is characterised by clear, straw‑coloured fluid collecting in a tissue or cavity. This fluid is an exudate if due to inflammation (slightly protein‑rich) or a transudate if due to non‑inflammatory causes, but in pathology exams, serous inflammation refers to the inflammatory exudate. The epithelium remains intact, and there is minimal cellular content, especially few neutrophils. It reflects mild injury and resolves easily without scarring.
Morphology - fibrinous inflammation
Associated with increased vascular permeability.
Exudate contains large molecules (fibrinogen).
Form fibrin which is deposited in the extracellular space.
Characteristic of inflammation of meninges, pericardium, and pleura.
Fibrinous exudate may be removed by fibrinolysis and macrophages.
If fibrin is not removed, the exudate may be converted to scar tissue (organisation).
Scar may compromise the function of the tissue / organ.
Fibrinous inflammation is characterised by the deposition of fibrin in the extracellular space as a result of increased extravascular permeability.
Fibrinous inflammation is characterised by fibrin‑rich exudate deposited on serosal surfaces. Microscopically, you see eosinophilic fibrin threads forming a meshwork; grossly, the surface appears dull, rough, and “bread‑and‑butter”‑like. This occurs when the injury is more intense, allowing large molecules like fibrinogen to escape. If the fibrin is not removed by macrophages, it can organise into scar tissue, leading to adhesions.
Morphology - supparative (puralent) - PUS
Purulent exudate composed of neutrophils, liquefactive necrosis, and edema.
Indicative of prominent cellular components.
Associated with pyogenic bacterial infection (staphylococci).
Heme of neutrophil MPO gives the exudate its characteristic colour.
Abscesses
Focal collections of purulent inflammatory tissue buried in a confined space.
Supparative inflammation is characterised by the production of pus, an inflammatory exudate composed of neutrophils and dead cells.
Suppurative inflammation occurs when massive neutrophil recruitment leads to the accumulation of pus, a mixture of living and dead neutrophils, necrotic cells, and bacteria. The tissue undergoes liquefactive necrosis, producing a soft, semi‑fluid mass. When this process becomes localised and walled off, it forms an abscess. Grossly, the area appears swollen, yellow‑white, and often fluctuant. Microscopically, you see sheets of neutrophils, necrotic debris, and sometimes a surrounding rim of dilated vessels and fibroblasts if chronicity develops.
Morphology - ulceration
Necrosis and inflammation on or near the surface.
Mucosa of mouth, GIT, genitourinary tract.
Skin - lower extremeties with poor circulation.
Shedding of inflamed necrotic tissue.
Ulceration occurs when severe inflammation causes necrosis of the epithelium, exposing the underlying connective tissue. The surface is lost, forming a sharply demarcated defect. The base of the ulcer contains granulation tissue, fibrin, and neutrophils in acute stages; with time, macrophages, lymphocytes, and fibrosis appear, reflecting chronicity. Ulcers can bleed, become infected, or heal with scarring depending on depth and duration.
Chronic inflammation
Primary chronic inflammation: injury that involves chronic inflammation without an initial acute inflammatory response.
Persistent infections.
Immune mediated disease (hypersensitivity)
Prolonged exposure to toxic agents.
Primary granulomatous.
Chronic inflammation
Prolonged inflammation (weeks, months, years).
Inflammation + injury + healing.
Characterised by:
Mononuclear cell infiltrations.
Tissue destruction.
Repair involving angiogenesis and fibrosis.
Chronic inflammation arises when the injurious stimulus persists or when the acute response fails to eliminate it. It is characterised by infiltration with mononuclear cells (macrophages, lymphocytes, plasma cells), ongoing tissue destruction driven by these cells, and attempts at healing through fibrosis and angiogenesis. Causes include persistent infections (e.g., TB), autoimmune diseases, prolonged exposure to toxic agents, and foreign bodies. Unlike acute inflammation, which is neutrophil‑rich and short‑lived, chronic inflammation is slow, destructive, and fibrotic, often leading to permanent tissue damage.
Examples of primary chronic inflammation
Rheumatoid arthritis
Chronic inflammation.
Autoimmune.
Dense inflammatory infiltrates (frequently forming lymphoid follicles) of CD4+ helper T cells, B cells, plasma cells, dendritic cells, and macrophages.
Rheumatoid arthritis is caused by a loss of immune tolerance, leading to autoantibody formation (RF and anti‑CCP) and chronic synovial inflammation driven by CD4⁺ T cells, macrophages, and cytokines such as TNF and IL‑1. The inflamed synovium becomes hyperplastic, forming a pannus that invades cartilage and bone, causing joint deformity, ankylosis, and loss of function. RA is symmetric, affects small joints (MCP, PIP), and has systemic features like fatigue, anaemia of chronic disease, nodules, serositis, and vasculitis. It is a classic example of chronic inflammation with ongoing tissue destruction and attempted repair.
As a consequence of unresolved acute inflammation….
Progression from an episode of acute inflammation.
Persistence of cause - interference with normal healing.
Peptic ulcer, chronic abscess, bronchiectasis, osteomyelitis.
Recurrent episodes of acute inflammation (chronic cholecystitis).
When acute inflammation fails to clear the offending agent, the persistent injury leads to ongoing neutrophil‑mediated damage, tissue necrosis, and activation of macrophages and lymphocytes, driving the transition to chronic inflammation. This chronic state results in fibrosis, scarring, loss of tissue function, and sometimes granuloma formation if the stimulus is difficult to eradicate. In some cases, unresolved acute inflammation leads to abscess formation, ulceration, or persistent exudates. Ultimately, unresolved acute inflammation becomes a self‑perpetuating cycle of injury and repair that can permanently alter tissue structure.
Chronic peptic ulcer
Associated with infection (H. Pylori) or NSAID use.
Acute inflammation with evidence of resolution interrupted by epithelial injury resulting in evidence of both acute and chronic inflammatory processes.
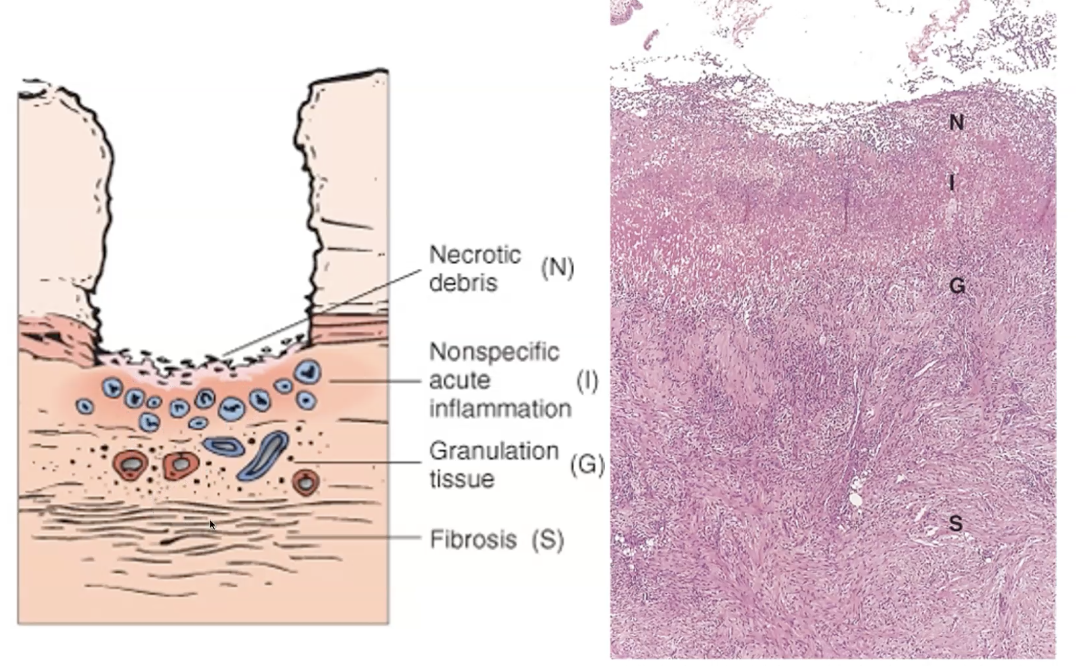
A chronic peptic ulcer forms when the mucosal defences of the stomach or duodenum are overwhelmed by acid and pepsin, leading to focal mucosal necrosis that extends into the submucosa or deeper. Because the injury persists, the ulcer develops a characteristic four‑layered morphology: a surface of necrotic debris, a layer of acute inflammation, a deeper zone of granulation tissue, and finally a fibrous scar at the base. Chronic ulcers have punched‑out edges, a clean base, and are surrounded by chronic inflammatory cells. Complications include bleeding, perforation, obstruction, and malignant transformation (rare in duodenal ulcers, possible in gastric ulcers).
Cells involved in chronic inflammation
Other cells involved in chronic inflammation
Eosinophils:
Parasitic infection and allergic reactions.
Granules release major basic protein toxic to parasites and causes epithelial cell necrosis.
Mast cells
Acute and chronic inflammation.
Early vascular change in acute inflammation (histamine, leukotrienes).
Allergic reaction (IgE).
Neutrophils
Although a hall mark of acute inflammation maybe present in some forms of chronic inflammation.
Chronic inflammation is characterised by infiltration of macrophages, lymphocytes, and plasma cells, with contributions from eosinophils, mast cells, and sometimes fibroblasts during repair. Macrophages are the central players: they secrete cytokines, present antigen, destroy tissue, and stimulate fibrosis. Lymphocytes (especially CD4⁺ T cells) orchestrate the immune response, while plasma cells produce antibodies. Eosinophils dominate in parasitic infections and allergy, and mast cells contribute to vascular changes and IgE‑mediated reactions. Together, these cells create a cycle of inflammation → tissue injury → repair, which defines chronic inflammation.
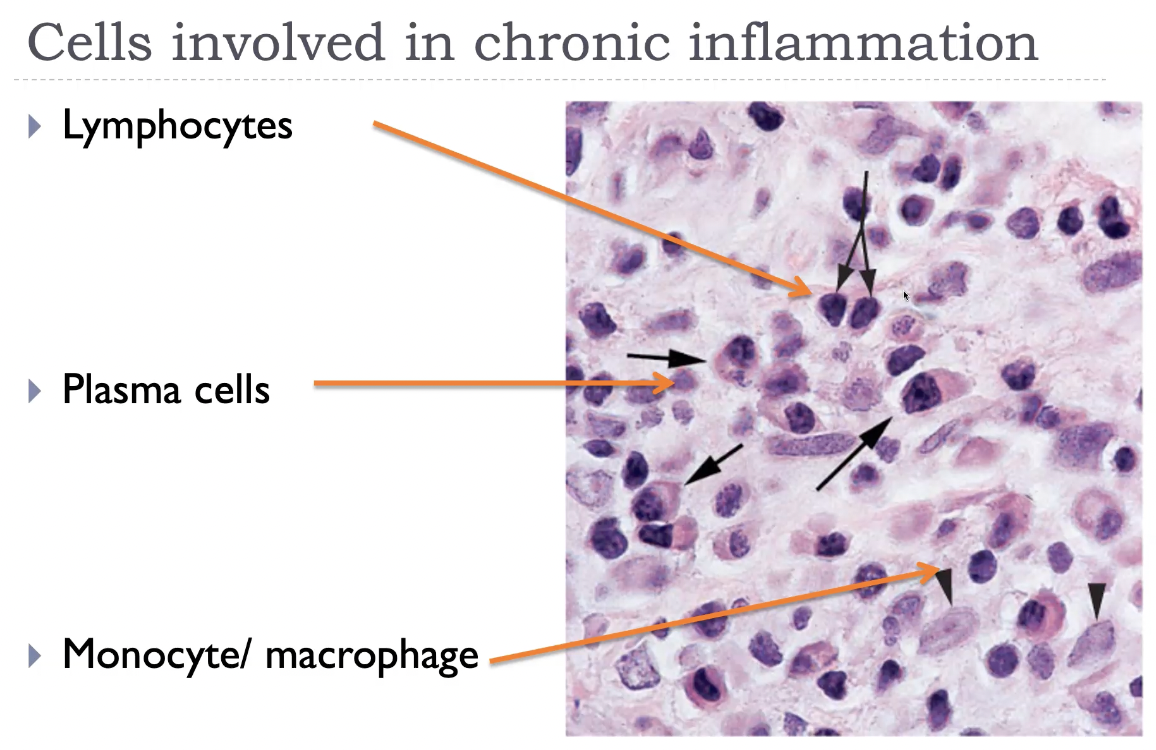
Monocyte / macrophages
Monocytes (immature mononuclear phagocytes) recruited and differentiate into macrophages in response to inflammation.
Tissue macrophages
Initiate acute inflammation.
Cytokines - prolong inflammatory response leading to chronic inflammation.
Monocytes and macrophages form the backbone of chronic inflammation. Monocytes circulate in the blood and migrate into tissues, where they differentiate into long‑lived macrophages under the influence of cytokines such as M‑CSF. Once activated, macrophages become the dominant effector cells, driving both tissue destruction and repair. Through classical (M1) activation, they release TNF, IL‑1, ROS, NO, and proteases, sustaining inflammation and damaging surrounding tissue. Through alternative (M2) activation, they secrete IL‑10 and TGF‑β, promoting healing and fibrosis. Macrophages also act as antigen‑presenting cells, activating T lymphocytes, which in turn further stimulate macrophages—creating a self‑perpetuating cycle characteristic of chronic inflammation. Their ability to phagocytose debris, orchestrate immune responses, and induce fibrosis makes them the central regulators of long‑standing inflammatory disease.
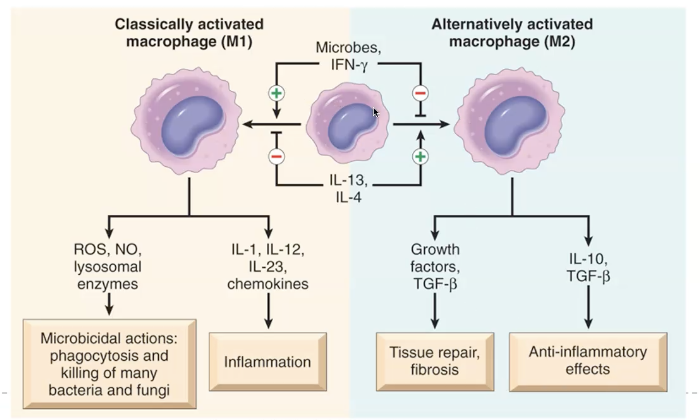
Tissue macrophage activation
M1 classical activation by microbial products and IFN-Y.
Microbicidal processes and inflammation.
M2 alternative activation by other cytokines (IL-4 and IL-13)
Principle role in tissue repair.
Tissue macrophages
Ingest and eliminate microbes and dead tissues.
Initiate the process of tissue repair.
Display antigens to T-lymphocytes and respond to signals from T cells.
Die or 'wander off' in the absence of inflammatory stimulus.
Tissue macrophages become activated when they encounter microbial products, cytokines, or signals from injured tissue. Classical (M1) activation, driven by IFN‑γ and microbial molecules like LPS, produces a highly pro‑inflammatory macrophage that releases TNF, IL‑1, ROS, NO, and proteases, sustaining inflammation and causing tissue injury. In contrast, alternative (M2) activation, driven by IL‑4 and IL‑13, generates macrophages that secrete IL‑10 and TGF‑β, promoting tissue repair, angiogenesis, and fibrosis. Activated macrophages also present antigen to T cells, which in turn further activate macrophages, creating a self‑reinforcing loop that defines chronic inflammation. In short: M1 macrophages kill; M2 macrophages heal.
Lymphocytes
Mobilised by immune stimulus (infection) and non-infection mediated inflammation.
Recruited into peripheral tissues.
B-lymphocytes develop into plasma cells and secrete antibodies.
T-lymphocytes:
CD4+ T-lymphocytes become activated and secrete cytokines.
CD8+ T-lymphocytes become activated and cytotoxic effector cells.
Lymphocytes are central regulators of chronic inflammation, working alongside macrophages in a self‑reinforcing loop. CD4⁺ T cells (especially Th1, Th2, and Th17 subsets) release cytokines that activate macrophages, recruit other leukocytes, and determine the type of inflammatory response. Th1 cells produce IFN‑γ to drive classical macrophage activation, Th2 cells promote eosinophilic inflammation and fibrosis, and Th17 cells recruit neutrophils and monocytes. B cells mature into plasma cells, producing antibodies that can form immune complexes or target self‑antigens in autoimmune disease. Lymphocytes accumulate in dense aggregates, sometimes forming lymphoid follicles, and their cytokines perpetuate tissue injury while simultaneously stimulating repair. Their presence is a hallmark of chronic inflammation and a key reason it becomes persistent and destructive.

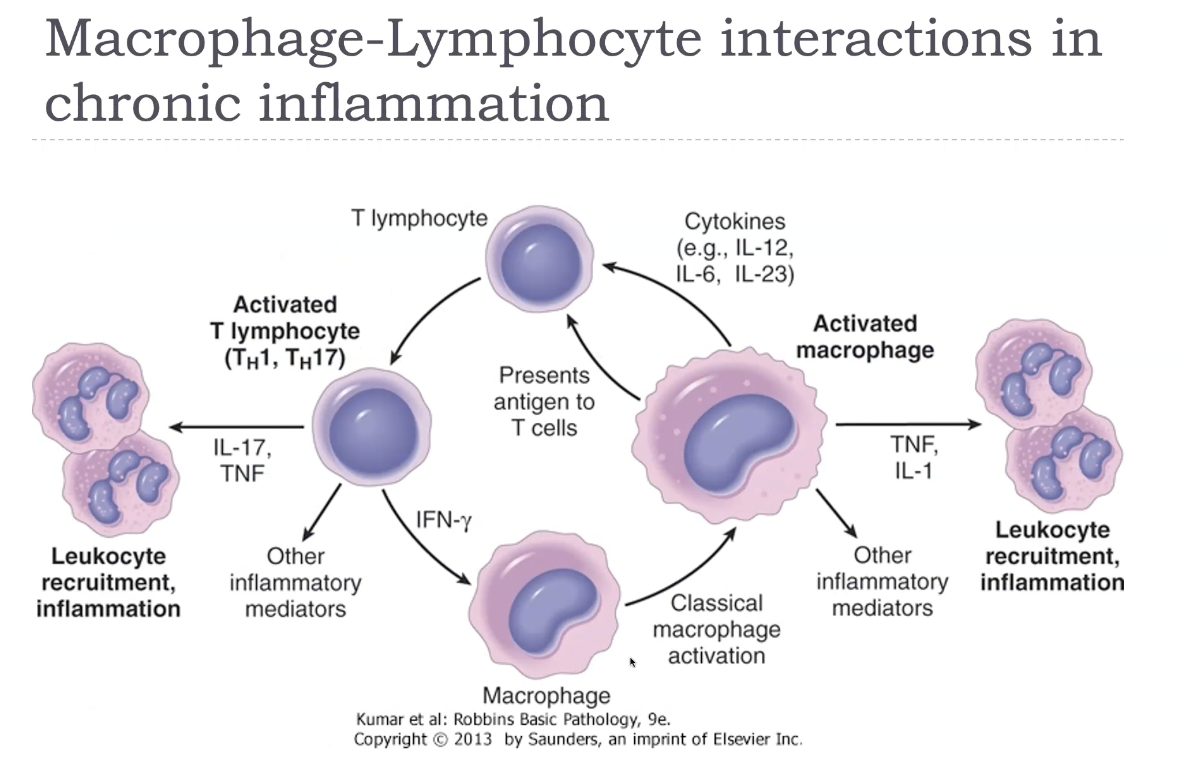
Macrophage-lymphocyte interactions in chronic inflammation
In chronic inflammation, macrophages and lymphocytes engage in a powerful bidirectional activation loop. Activated macrophages present antigen and secrete cytokines such as IL‑12, which stimulates CD4⁺ T‑cell differentiation—especially Th1 and Th17 subsets. These T cells then release cytokines like IFN‑γ (from Th1) and IL‑17 (from Th17), which further activate macrophages, enhancing their production of TNF, IL‑1, ROS, NO, and proteases. This cycle leads to persistent inflammation, ongoing tissue destruction, and fibrosis. Th2 cells can also influence macrophages by promoting M2 activation, driving repair and fibrosis through IL‑4, IL‑13, and TGF‑β. This macrophage–lymphocyte cross‑talk is the hallmark of chronic inflammation and explains why it becomes self‑sustaining and difficult to resolve.
CHRONIC INFLAMMATION - Morphology
Macroscopic features of chronic inflammation
Chronic ulcer
Chronic abscess cavity
Thickening of the wall of an organ.
Granulomatous inflammation
Fibrosis
Microscopic features of chronic inflammation
Inflammation:
Mononuclear cell infiltrations.
Macrophages, lymphocytes, plasma cells, eosinophils.
Injury:
Tissue destruction.
Persistent cause and mediators of inflammation - actions of inflammatory cells.
Repair:
Repair involving angiogenesis and fibrosis.
Proliferation of fibroblasts and endothelial cells.
Chronic inflammation is characterised morphologically by a dense infiltrate of mononuclear cells—primarily macrophages, lymphocytes, and plasma cells—surrounding areas of ongoing tissue destruction caused by these same inflammatory cells. The affected tissue shows evidence of repair, including proliferation of fibroblasts, collagen deposition, and angiogenesis, forming granulation tissue. Over time, this leads to fibrosis, architectural distortion, and loss of function. In some conditions, macrophages transform into epithelioid cells and fuse to form giant cells, producing granulomas, a specialised pattern of chronic inflammation.
Diffuse inflammation
Chronic bronchitis - exogenous toxin.
Bronchiectasis - persistent infection.
Asbestosis - exogenous toxin.
Ulcerative colitis - inappropriate immune response.
Chronic peptic ulcer - persistent infection.
Chronic cholecystitis - recurrent acute cholecystitis.
Rheumatoid arthritis - inappropriate immune response.
Atherosclerosis - endogenous toxin
Diffuse inflammation is characterised by widespread infiltration of inflammatory cells—usually mononuclear cells in chronic inflammation or neutrophils in acute inflammation—distributed uniformly throughout the affected tissue. Instead of forming a discrete focus, the inflammation blends into the surrounding tissue, often accompanied by diffuse oedema, vascular congestion, and tissue destruction. In chronic diffuse inflammation, there is also fibroblast proliferation, collagen deposition, and architectural distortion. This pattern is typical of diseases such as diffuse interstitial pneumonia, diffuse hepatitis, and diffuse myocarditis, where the entire organ shows inflammation rather than a focal lesion.
Histological features of peptic ulcer
Necrosis-fibrinoid debris.
Inflammation - neutrophils.
Granulation tissue - macrophages, endothelial cells, fibroblasts.
Scar - fibrous collagenous scar.
A peptic ulcer shows a sharply demarcated mucosal defect with a characteristic four‑layered histological appearance. The surface contains necrotic, eosinophilic debris, underlain by a band of acute inflammatory cells, mainly neutrophils. Beneath this lies granulation tissue composed of proliferating capillaries, fibroblasts, and chronic inflammatory cells. The deepest layer is a dense fibrous scar, which may distort the underlying muscularis propria. Surrounding mucosa often shows chronic gastritis. This layered pattern reflects ongoing injury from acid and pepsin alongside attempts at healing.
Asbestosis
Diffuse pulmonary interstitial fibrosis.
Contraction - enlarged air spaces within thick fibrous walls.
Fibrous thickening of visceral pleura.
Asbestosis is characterised by diffuse interstitial fibrosis, predominantly in the lower lobes and subpleural regions, caused by the persistent inflammatory response to inhaled asbestos fibres. Macrophages attempt to phagocytose the fibres but cannot degrade them, leading to chronic macrophage–lymphocyte activation, release of fibrogenic cytokines (especially TGF‑β), and progressive scarring. The hallmark histological feature is the asbestos (ferruginous) body—a golden‑brown, beaded, dumbbell‑shaped fibre coated with iron‑containing protein. Over time, the lung becomes stiff, honeycombed, and functionally restricted. Asbestosis increases the risk of lung carcinoma and is strongly associated with malignant mesothelioma.
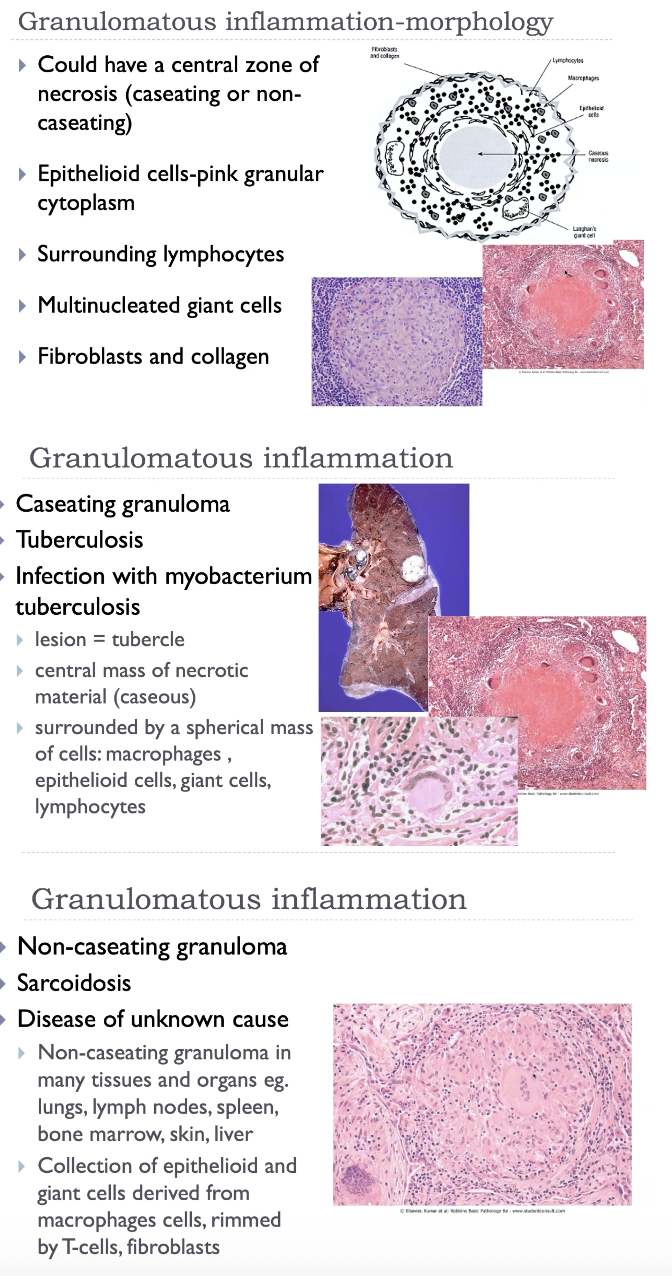
Granulomatous inflammation
Aggregates of activated macrophages
Epithelioid cells - epithelial-like appearance (large, pink, flat).
Giant cells - fusion of macrophages in response to chemical mediators (contain many nuclei).
T-lymphocytes (causing chronic macrophage activation).
Rim of fibroblasts and connective tissue.
Central zone of necrosis.
'Walls off' offending agent-defense mechanism.
Subsequent fibrosis may cause dysfunction.
Granulomatous inflammation is characterised by the formation of granulomas, which are organised collections of epithelioid macrophages—activated macrophages with abundant pink cytoplasm—often surrounded by a collar of lymphocytes. These epithelioid cells may fuse to form multinucleated giant cells. Granulomas arise when the immune system cannot eliminate a persistent stimulus, such as Mycobacterium tuberculosis, fungi, parasites, foreign bodies, or in immune‑mediated diseases like sarcoidosis and Crohn disease. Depending on the cause, granulomas may be caseating (necrotic, cheese‑like centre, typical of TB) or non‑caseating (sarcoidosis). Over time, granulomas may undergo fibrosis, contributing to tissue dysfunction.
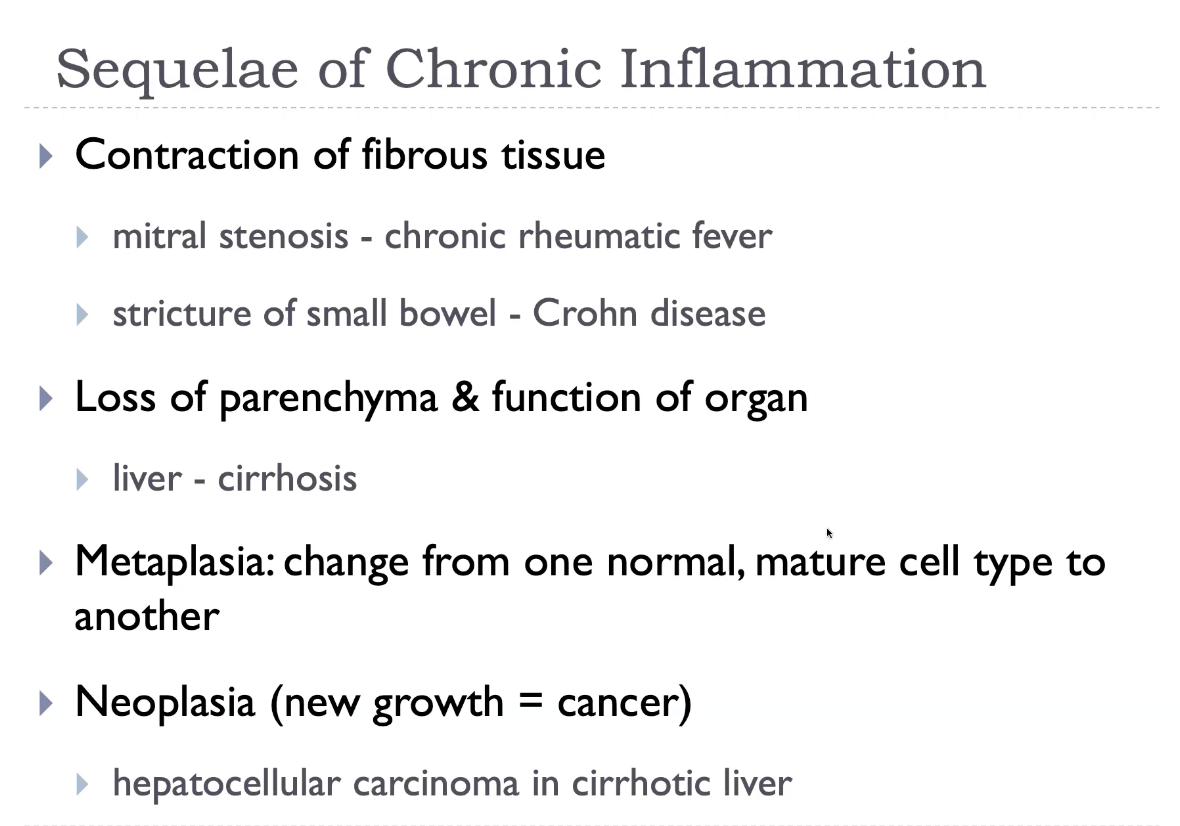
Sequelae of chronic inflammation
Chronic inflammation leads to progressive tissue destruction, followed by fibrosis and scarring, which distort normal architecture and impair organ function. Persistent cytokine release drives angiogenesis, fibroblast proliferation, and collagen deposition, resulting in stiff, poorly functioning tissue. Chronic inflammatory states may also produce granulomas, metaplasia, and even dysplasia, increasing the risk of malignancy in some organs. Systemically, chronic inflammation contributes to anaemia of chronic disease, cachexia, and amyloidosis. These sequelae reflect the imbalance between ongoing injury and incomplete repair.