Gene Technology and Genomics
1/420
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
421 Terms
Genetic Engineering
Methods of recombinant DNA technology (or gene cloning), where DNA molecules from two or more sources are combined either within cells or in vitro and are then inserted into host organisms in which they are able to propagate.
Gene Technology
Any technique for the modification of genes or other genetic material, but does not include sexual reproduction, homologous recombination or any other techniques specified in the regulations.
Genetically Modified Organism (GMO)
An organism that has been modified by gene technology; or an organism that has inherited traits from an organism that occurred in the initial organism because of gene technology but does not include:
a human being
an organism declared by the regulations not to be a GMO
Two Techniques Before Recombinant DNA Technology
Agricultural genetic engineering and classical genetics.
Agricultural Genetic Engineering
Selective breeding for domestication of crops and livestock which was imprecise and created desirable recombinants slowly.
Classical Genetics Genetic Engineering
Make random mutations using chemicals or radiation
First characterise phenotype, then associate gene
Successful for limited number of traits and organisms
Successful for bacteria and “model” higher organisms
What does the Gene Technology Act of 2000 aim to do?
Protect the health and safety of people, and the environment, by identifying risks posed by or as a result of gene technology, and by managing those risks through regulating certain dealings with genetically modified organisms (GMOs).
The OGTR
Gene technology regulation is administered by the Gene Technology Regulator supported by the Office of the Gene Technology Regulator (OGTR).
What does the OGTR regulate?
Live and viable genetically modified organisms, research, production, culture, breeding, import, and transport.
What doesn’t the OFTR regulate?
Intellectual property
Cost/benefit considerations
Trade and market impacts
Animal welfare considerations
Exempt Dealings
Very low risk, contained, involving well understood organisms and no intentional release into environment
Notifiable Low Risk Dealings (NLRDs)
Low risk, conditions specified in regulations observed, including use of certified physical containment (PC) facilities (e.g,. PC1, PC2, etc.).
Who Needs to Apply to Import or Use (Deal with) a GMO?
Exempt dealings – low risk – do not need approval
All other dealings can be conducted by Authorised Organisations and must have access to an Institutional Biosafety Committee (IBC) (includes experts and an external layperson). Individuals prepare applications that are evaluated by IBC.
Regulation of CRISPR-mediated Gene Editing
Individual countries providing rulings on whether to regulate GMOs, there is no international consensus.
The most contentious areas of debate revolve around agriculture and human germline editing.
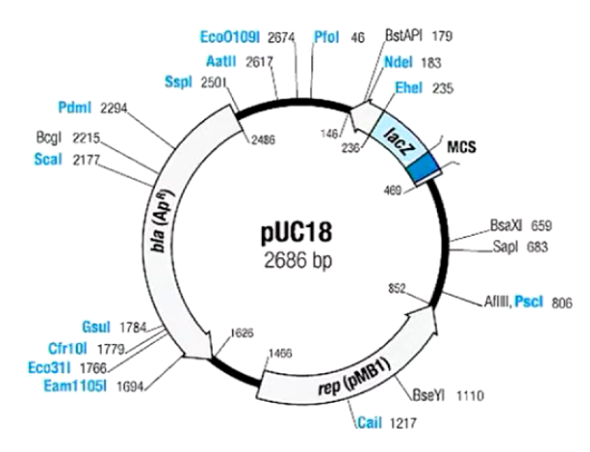
Plasmid Cloning Vector
Often small in size and are found in large numbers in a bacterium to allow for optimal cloning. They have 3 key features: an origin of replication, a selectable markers such as an antibiotic resistant gene, and a multiple cloning site (ex. lacZ) where the restriction enzyme cutting sites are found.
Odd End/Directed Cloning
Used when correct orientation of the gene being added to the plasmid is crucial
Cut it with two different restriction enzymes so that the overhangs don’t match up with each other and re-ligation cannot occur
Blue/White Screening
The E.Coli LacZ gene encodes the enzyme beta-galactosidase. When bacteria with the LacZ gene are grown on agar containing X-gal, cleavage of the X-gal by the enzyme causes a blue product to form. Many restriction enzymes cut within the LacZ gene so transformed bacteria colonies appear white.
Sources of DNA for Experiments
Genomic DNA, cDNA, and synthetic DNA
Why would synthetic DNA be used?
Optimised codon usage for host can increase expression levels
Often a lot cheaper and faster especially if the gene of interest is expressed at low levels so creating cDNA is difficult
TA Cloning
A type of gene cloning technology that uses Taq polymerase which during PCR will amplify the DNA being cloned but will add a 3’-A overhang at the end. The ends of this PCR product will hybdridise to linearised vector DNA possessing 3’-T overhangs, and is then ligated.
How are the T vectors for TA cloning prepared?
By linearisation with a blunt RE followed by treatment with enzyme terminal transferase in the presence of ddTTP (dideoxythymidine triphosphate).
Features of TA Cloning
Enables cloning without restriction enzymes
It is non-directional
Many pre-prepared “T vectors” of various types available commercially
TOPO TA Cloning
A type of gene cloning technology that uses the enzyme DNA topoisomerase I which acts as both the restriction enzyme and ligase. After Taq polymerase has amplified the PCR product DNA topoisomerase I Recognizes the sequence 5´-(C/T)CCTT-3´ and forms a covalent bond with the phosphate group attached to the 3´ thymidine to create a ds break. Released upon re-ligation of DNA ends.
Features of TOPO TA Cloning
Faster (5 minutes at room temperature)
More efficient (up to 95%)
It is non-directional
3 Ways to Determine the Direction of a Plasmid Insert
PCR, restriction digestion, and sequencing
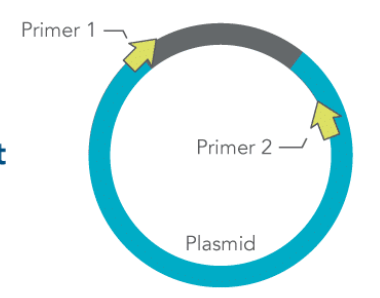
How can PCR be used to determine the direction of a plasmid insert?
Use a primer that’s located inside the inserted gene and one that is located on the normal plasmid
A PCR product will be obtained if it is in the correct orientation
How can restriction digestion be used to determine the direction of a plasmid insert?
Use a restriction enzyme that cuts once inside the insert (ideally off-center, such as at 1/3 of the length) and once in the adjacent vector backbone
The same enzyme cuts at the same locations relative to the backbone, but due to the flipped insert, it produces different sized fragments
Restriction-Enzyme Cloning
Used to create new combinations of DNA sequences by digesting DNA from different sources and joining them with DNA ligase
Cuts a target gene and a plasmid vector with specific, matching restriction enzymes to create complementary "sticky ends"
Methods of Gene Transfer for Plant Transformation
Agrobacterium transformation
Electroporation of protoplasts
Microprojectile bombardment
Microinjection
Viral infection
2 Outcomes of Gene Transfer
Transient – no incorporation of exogenous DNA into the genome
Stable – incorporation into genome
Agrobacterium tumefaciens
Bacterial plant pathogen found in soil that causes Crown gall disease in plants. It naturally transfers DNA to plants.
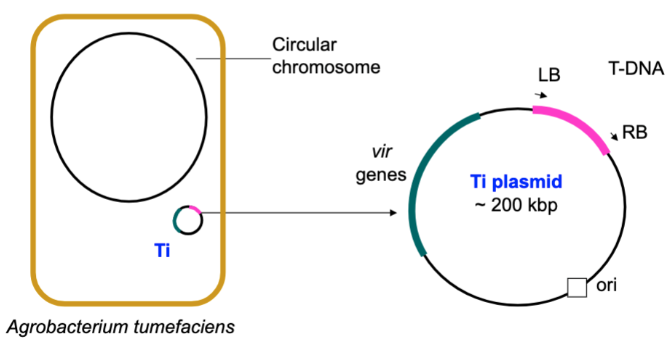
Ti (tumour-inducing) Plasmid
Found in Agrobacterium and has genes involved in crown gall disease.
T-DNA transfer into plants
Transformed cells start making cytokinins and auxins – these cells de-differentiate, divide and form tumours. De-differentiated cells make opines - specific nutrients for the bacterium.
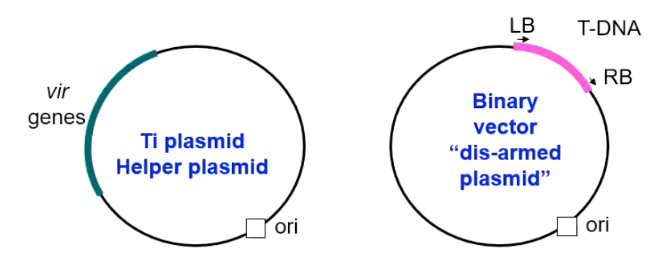
T-DNA Binary Vector System
A two-plasmid system used for Agrobacterium-mediated plant transformation, consisting of a small, manageable vector carrying the gene of interest and a separate "helper" plasmid containing the vir genes. It enables efficient gene transfer by separating essential functions, allowing for easier manipulation and higher transformation frequencies.
T-DNA Binary Vectory System Process
Generate DNA plasmid clone in E. coli
Introduce the plasmid into Agrobacterium containing a modified Ti plasmid
Infect plant tissue with engineered Agrobacterium
Select for transformed plants
Transgene insertion…
Is largely random
heterochromatin (not transcriptionally active)
euchromatin (strongly expressed)
What might random transgene insertion cause?
Variation of expression exhibited by identical transgenes that insert into different regions of a genome
level of expression
tissues where transgene is expressed
T-DNA may have knocked-out an existing gene that results in a phenotype
2 Ways to Transfer Genes Into Plants Using Agrobacteria
By tissue culture which works for most plants or by floral dip which is simpler (doesn’t need aseptic technique) but doesn’t work for most plants.
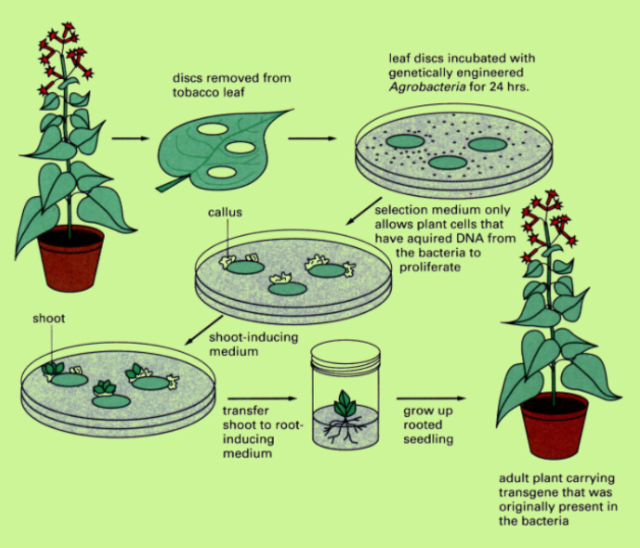
Process of Tissue Culture Gene Transfer
Can grow on Kanamycin to select for transformed plant cells. Have to include nptll gene in plasmid construct
Discs removed from leaf
Leaf discs are incubated with genetically engineered Agrobacteria for 24 hours
Selection medium only allows plant cells that have acquired DNA from the bacteria to proliferate
They are transferred to a shoot-inducing medium
The shoots are transferred to a root-inducing medium
The plant is grown up and the result is an adult plant carrying the transgene
Process of Floral Dip Gene Transfer
Transform Agrobacterium with T-DNA
Dip Arabidopsis flowers in Agrobacterium
Let plants self-fertilise
Harvest seed
Select KanR transformants or select RFP positive seed
Transplant to soil
What is T-DNA used for?
Used to generate mutants
Used to find out where and when genes are expressed
Analysis of gene function (increase or decrease gene expression)
Why are plants manipulated for agriculture?
Improve crop resistance to disease
Improve crop yields
Improve crop nutritional value
Advantages of Producing Transgenic Plants
Rapid transfer of traits between plants relative to breeding
Transfer traits between plants that don’t naturally hybridise
Develop plants with novel traits from other organisms
Alter pre-existing traits to obtain improved products
Disadvantages of Transgenic Plants
Cultured plant cells are genetically unstable
Genetic diversity of crop plants may be further reduced
3 Main GMO Safety Concerns
tendencies to be toxic or to provoke allergic reaction (allergenicity)
gene transfer to other species
outcrossing of transgenic plants to wild species
Flavr Savr Tomato
Tried to improve safe transport of ripe fruit by preventing the softening of the fruit. But shelf life was not extended and it could not be harvested when ripe and be shipped safely.
Main Sources of GM Foods in Australia
imported GM soya
cottonseed oil made from GM cotton
imported GM corn
imported GM sugar beet
grown in Australia GM canola and safflower
What regulates GM foods and ingredients in Australia?
FSANZ
What does FSANZ investigate?
Nutritional content, toxicity, tendency to provoke any allergic reaction, stability of the inserted genetic material, whether there is any nutritional deficit or change in the GM food or ingredient, and any other unintended effects of the gene insertion. A GM food will only be approved for sale if it is assessed as being safe and as nutritious as its conventional, non-GM counterpart.
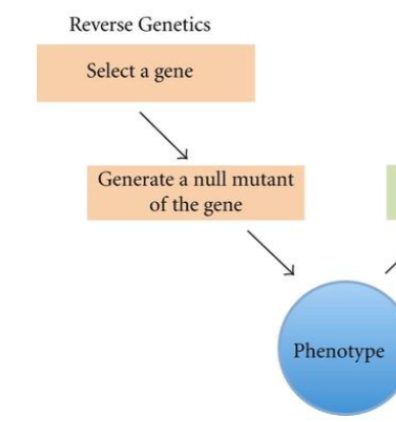
Reverse Genetics
Determines a gene's function by starting with a known DNA sequence and analyzing the phenotype caused by deliberately altering or mutating that gene.
Knocking Out
DNA (eg coding sequence) mutation (loss-of-function mutation)
Knocking Down
Reduced expression - reduction in transcription or translation of a gene (stable or transient).
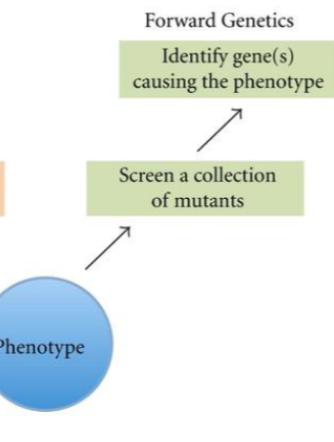
Forward Genetics
Aims to identify the sequence that underlies a specific phenotype. Induce random mutations and identify phenotype of interest. Determine the gene causing the phenotype.
Epistasis
Epistatic gene masks the expression of the hypostatic gene.
Transient Assays
Give rapid functional readouts; stable mutants require time to generate. Include non-specific mutagenesis methods such as chemical (e.g., EMS) and radiation mutagenesis which cause random mutations across the genome.
Reporter-Based Hypothesis Testing
Firefly luciferase (fLUC) provides a quantifiable output for promoter or gene-regulation assays. The reporter gene commonly used is Firefly luciferase, which produces light when it is expressed. By measuring the amount of light, researchers can quantitatively determine how strongly the promoter or regulatory element is activating gene expression.
Targeted Mutagenesis Technologies
Engineered nucleases: ZFN, TALEN, CRISPR-Cas9.
Enable precise DNA modifications via NHEJ (indels) or HDR (templated repair).
Ethyl methanesulfonate (EMS) Mutagenesis
EMS is an organosulfur compound and is most commonly used chemical mutagen in experimental genetics. It produces random point mutations by nucleotide substitution.
How does EMS introduce mutations?
EMS ethyl group reacts with guanine, forming the abnormal base O6-ethylguanine.
During DNA replication, DNA polymerases place thymine, instead of cytosine, opposite O6-ethylguanine. Subsequent rounds of replication lead to an A:T pair (a transition mutation).
Ionizing Radiation Mutagenesis
exposing cells or organisms to high-energy radiation such as X-rays or gamma rays. This radiation damages DNA (e.g., causing strand breaks or base changes), which can lead to mutations when the DNA is repaired.
Can cause whole chromosomes to fuse with each other
Process of Ionizing Radiation Mutagenesis
Interaction of gamma rays with water molecules generates free radicals
Free radicals and gamma rays induce DNA breaks
Changes in DNA codons and errors are incorporated into the DNA
3 Methods of Targeted Mutagenesis Technologies
Zinc Finger Nucleases, TALENSs, and CRISPR/Cas9
NHEJ-Mediated Repair
Process of DNA repair in response to double strand breaks that directly ligates broken DNA ends without requiring a homologous template, often causing small insertions or deletions (indels).
Homology Directed Repair (HDR)
A precise DNA damage repair mechanism used in genome editing to fix double-strand breaks using a homologous template. Can be used to add a donor template into the DNA.
Zinc Finger Nucleases
Engineered, site-specific restriction enzymes that create double-strand breaks and are created by fusing a customizable zinc-finger DNA-binding domain to a DNA-cleavage domain.
2 Domains of Zinc Finger Nucleases
DNA-binding Zinc Finger motif (3-6 motifs per ZFN)
Variable Zinc finger motifs adaptable to DNA structure and nucleotide binding (1 motif per three nucleotide)
Nuclease domain from the Fok1 restriction enzyme: Two monomers required for Fok1 to cleave DNA (non-sequence specific)
Drawbacks of Zinc Finger Nucleases
Challenges in creating specific DNA binding domains
No open-source collection of 64 Zinc Fingers that covers all possible combinations of 3 bp triplet sites
Expensive
TALENS
Transcription activator-like effector nucleases which are highly precise, customizable DNA-cutting enzymes used for genome editing that use TALE DNA binding domains.
What are TALE DNA Binding Domains
33-35 amino acid repeats (each individually developed to recognize a single base pair)
Specificity created by a repeat variable di-residue (RVD) located at position 12 and 13 of each aa repeat
RVD’s: Adenine (NI), Cytosine (HD), Thymine (NG), Guanine (NH), AG (NN), ACGT (NS)
CRISPR-Cas9
It uses a guide RNA (about 20 nucleotides) to direct the Cas9 enzyme (molecular scissors) to a specific DNA sequence, creating a double-strand break to fix, remove, or alter genetic material
The only thing that needs to be reprogrammed is the guide RNA
PAM Site
The protospacer adjacent motif used for CRISPR which cuts next to the PAM sequence
A short (2-6 base pair) DNA sequence immediately following the target DNA (protospacer) that is essential for Cas9 enzyme binding and cleavage. It distinguishes target DNA from self-DNA
Often NGG
crRNA
CRISPR-associated RNA which is the guide RNA
tracrRNA
Trans-activating RNA that is a scaffold that allows for the binding of the guide RNA to the Cas9 protein.
sgRNA
Single guide RNA which is an RNA segment with the guide and scaffold connected in a single long RNA.
CRISPR Innovations
Easily target precise locations on the DNA.
Can introduce errors to switch off protein function.
Introduce new DNA into the genome.
Guide other functional elements to precise locations on the DNA.
CRISPR Therapeutic to Treat Sickle Cell Anemia
CRISPR has been used in a therapy, CTX001, to make a small edit in a cis-acting element (GATA1-binding site) in the BCL11A promoter.
Mutation of the GATA1-binding site leads to reduced suppression of the fetal, gamma globin in adult erythroid (red blood) cells
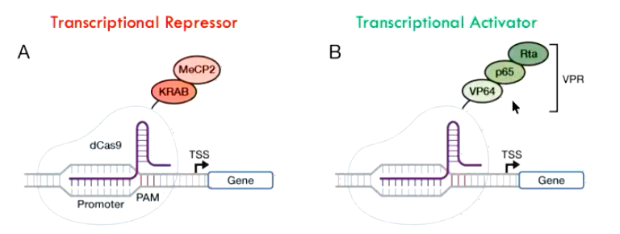
Disabled Cas9
Can be disabled to guide transcriptional repressors or activators to a gene of interest. These then cause suppression or over-expression of a gene.
Base Editors
Made by fusing a catalytically impaired "dead" Cas9 (dCas9) or nickase Cas9 (nCas9) to a deaminase enzyme. Cause the irreversible conversion of one DNA base pair into another without generating double-stranded DNA breaks (DSBs).
3 Types of Base Editors
Cytosine base editors (CBEs), adenine base editors (ABEs), C-to-G base editors (CGBEs).
Cytosine Base Editors (CBEs)
Install C•G-to-T•A point mutations using Cas9 nickases or dCas9 fused to cytidine deaminases and uracil glycosylase inhibitor domains (UGI).
Process of CBEs
CBEs bind to a target DNA sequence and form a single-stranded R-loop.
The cytidine deaminase, targets an exposed cytosine and removes its amine group, converting it into uracil.
UGIs inhibit uracil glycosylases, which would excise the uracil base to form an abasic site. Abasic sites lead to indel formation.
The Cas9 domain nicks the non-deaminated strand, which directs DNA repair to install an adenine opposite the uracil as the nicked strand is remade.
Prime Editors
Consist of a Cas9 nickase domain fused to a reverse transcriptase domain.
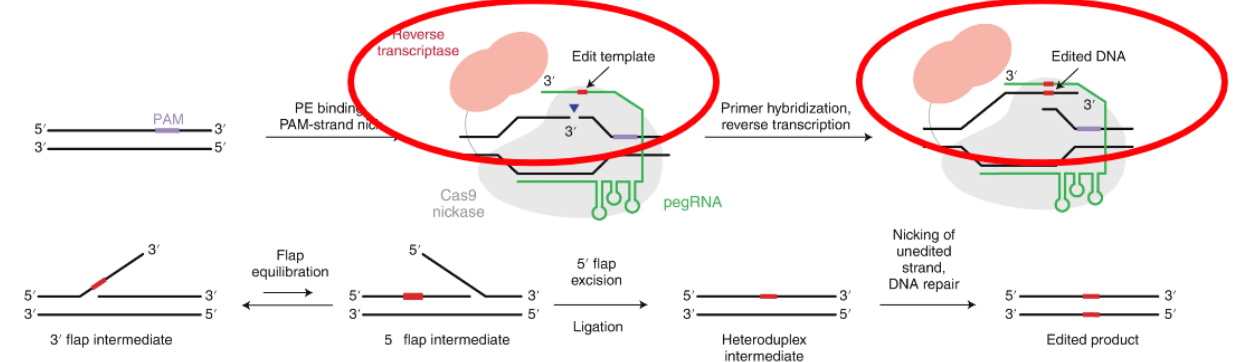
Prime Editors Process
The prime editing guide RNA (pegRNA) guides the prime editor to its genomic DNA target and also encodes the desired edit.
After nicking the PAM-containing strand, the DNA 3ʹ end hybridizes to the pegRNA extension to form a primer–template complex.
The reverse transcriptase domain then copies the template from the pegRNA extension into the genomic DNA directly, adding the edited sequence to the target locus.
The product of reverse transcription, an edited 3ʹ flap, can then incorporate into the DNA duplex by competing with the original and redundant 5ʹ flap sequence.
After 5ʹ flap excision and ligation of the edited strand, the non-edited complementary strand is replaced by DNA repair using the edited strand as a template.
Sanger Sequencing
A type of shotgun sequencing that sequences short stretches of DNA (~800-1,000 bp) and is cheap if only interested in a few genes. For whole genomes it is slow and expensive at scale.
dNTP vs ddNTP
dNTP: A generic term referring to the four deoxyribonucleotides: dATP, dCTP, dGTP and dTTP
ddNTP: modified with fluorescent tag and no hydroxyl group to prevent addition of next dNTP. ddNTPs lack both the 2’-OH and 3’-OH groups.
Ratio of dNT:ddNTP
The ratio of dNTP:ddNTP varies from around 10:1 to 300:1, depending on desired read length, buffer conditions, the polymerase used, and the electrophoresis conditions.
Sanger Sequencing Process
Design primers to bind near sequence of interest
Extension, using mix of dNTPs (normal nucleotides) and fluorescently labelled ddNTPs
Incorporation of ddNTP will stop extension and this will be labelled
Dye-labeled segments are applied to a capillary gels and subjected to electrophoresis
Chromatogram will show the different nucleotides and the order that they appear
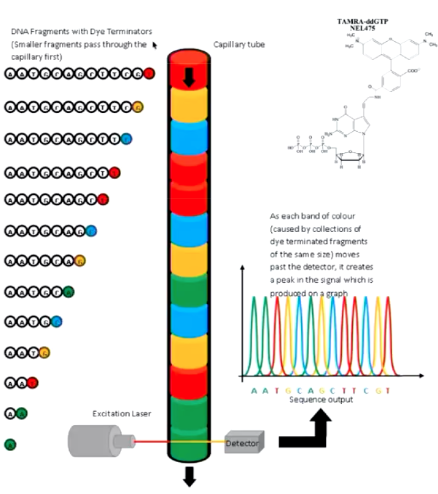
Sanger Sequencing Capillary
Separate DNA fragments of varying size using resin in a capillary tube. Still regularly used for small scale sequencing used in molecular biology such as cloning, vector construction, and PCR verification. The output is the sequence call is based on fluorescence peak size and its shape.
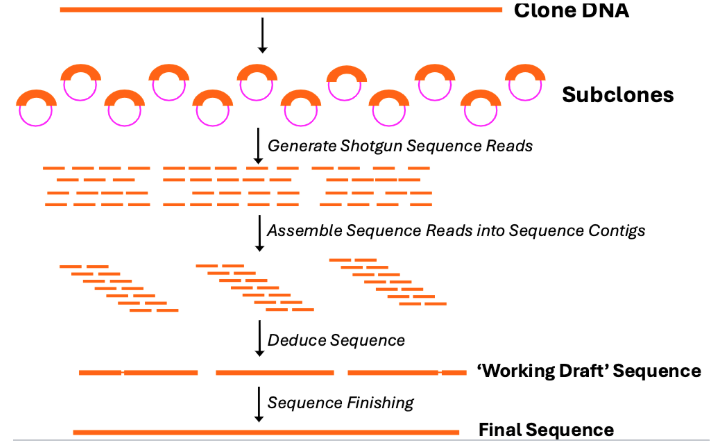
Shotgun Sequencing
A laboratory technique for determining the DNA sequence of an organism’s genome (or part of the genome). The method involves randomly breaking up the DNA into small fragments that are then sequenced individually. A computer program looks for overlaps in the DNA sequences, using them to reassemble the fragments in their correct order to determine the sequence of the starting DNA.
Next Generation Sequencing
Sequences many different strands at the same time which can be pieced together. Do not need information about sequence and is useful for larger scale applications such as RNA or genome sequencing.
Next Generation Sequencing Types
Includes pyrosequencing (454), semiconductor sequencing (Ion Torrent), and reversible chain-termination sequencing (Illumina).
DNA Sequence Assembly
Short sequence reads need to be aligned to create a full sequence
Contiguous sequences (“contigs”) are assembled from overlapping individual reads in a 5’-3’ orientation
Contigs are then arranged into longer, scaffold sequences
The scaffolds are subsequently arranged into putative chromosomes using information from a reference genome of a closely related species.
N50
The length of the shortest read that represents at least 50% of the total bases. A higher N50 indicates better, more contiguous genome assembly.
Oxford Nanopore
Solves the issue of repetitive sequences
A strand of DNA is passed through a nanopore
The magnitude of current is changed as the bases G, A, T and C pass through the pore in different combinations
N50 ranging from around 30kb to over 100kb.
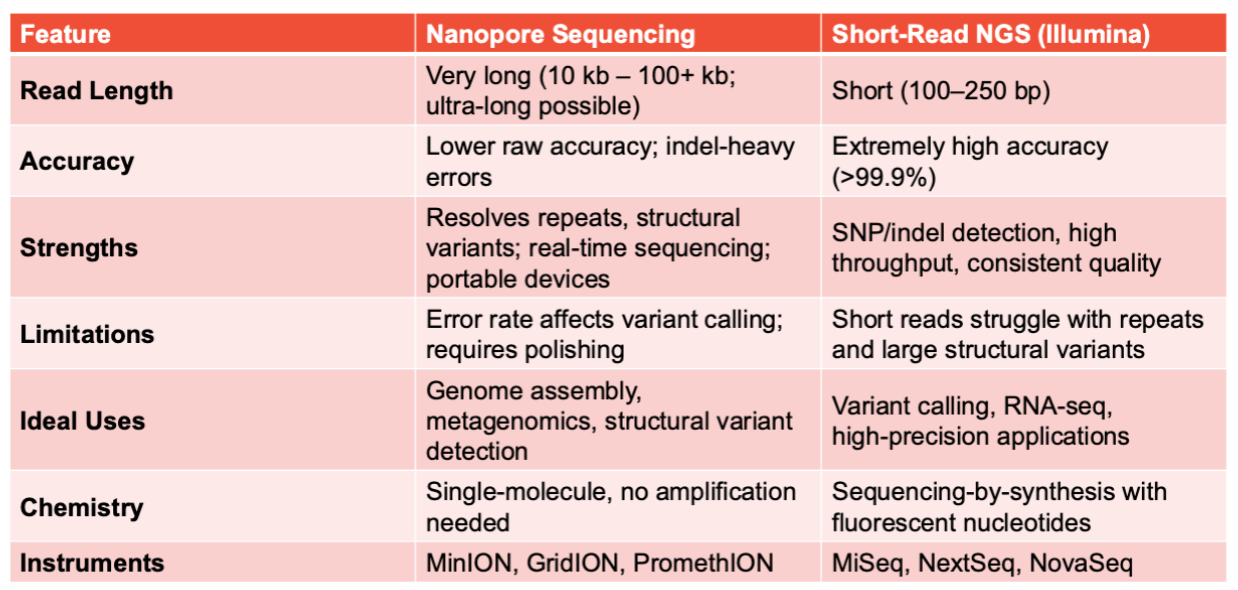
Read Length and Accuracy
Longer reads allows coverage over repetitive regions
Single molecule sequencing allows read lengths over 10 kB. But has a higher raw error rate (indel heavy errors) and base level comparisons and SNP/indel detection are less reliable (heavy polishing/hybrid approaches needed)
Comparison of Nanopore and Short-Read NGS
Sequence Costs…
Decline but interpretation costs rise. It is possible to quickly (re)sequence large complex genomes, assuming you have sufficient computing capability to process the data.
GenBank
National Institute of Health (NIH) genetic sequence database
Publically available annotated sequences
Sequence Database (number of bases) doubles in size every 18 months
Max Score in BLAST
Highest alignment score calculated from matches and mismatches found in alignments between the query sequence and the database, indicating the strength of the best match