DAPAB W5D2: Curation of genotypes
1/29
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
30 Terms
Call rates (per SNP and per individual)
Call rate
Proportion of genotypes that are called (observed)
SNPs with low call rate may have poor genotyping quality
Call rates (per SNP and per individual)
Calling of SNP genotypes from SNP arrays (in diploids)
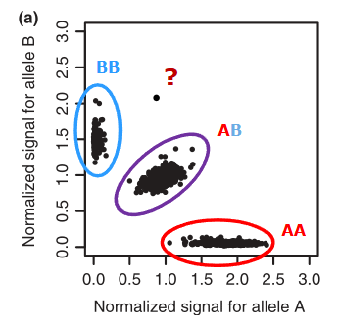
Call rates (per SNP and per individual)
Missing rate
Proportion of genotypes that are not called
Individuals with low call rate may have
Poor quality DNA
Contaminated DNA samples
Some (rare) disorder: chimera, aneuploidy
Call rates (per SNP and per individual)
Calculate proportion observed genotypes per SNP
Calculate proportion observed genotypes per individual
Determine call rate threshold
Typically, 95% or greater, depending on the genotyping quality
Plot the distribution of observed call rates
Determine a threshold based on discontinuity of the distribution
Minor allele frequency
SNPs have two alleles → two allele frequencies: p+q=1
MAF is the frequency of the least frequent allele; MAF=min(p,q)
Loci with (very) low MAF:
May not be informative
Removing too many may affect outcome subsequent analyses
May be due to genotyping erros
Determining a MAF threshold
Very low MAF may be due to genotyping errors
Expected error rate could be sensible MAF threshold
If an allele is observed more often, the probability that these are (only) due to genotyping errors decreases
So:
Decide how often an allele should be seen to be credible
Convert this number into a MAF threshold
MAF should always be >0
To at least remove non-segregating SNPs (with MAF=0)
Consider the number of genotyped individuals
BASE MAF on minimum number observations for an allele
Determing a MAF threshold - example
We have 1000 genotyped (diploid) individuals
A locus with
All animals having (true) genotype GG
A genotyping error rate of 0.1%Two individuals have a called genotype GA
For example:
MAF = 100%*(2/2000) = 0.1%
For the example:
Allele should be seen 5 times to be credible
100%/(5/2000)=-0.4%
Hardy-Weinberg equilibrium
Example
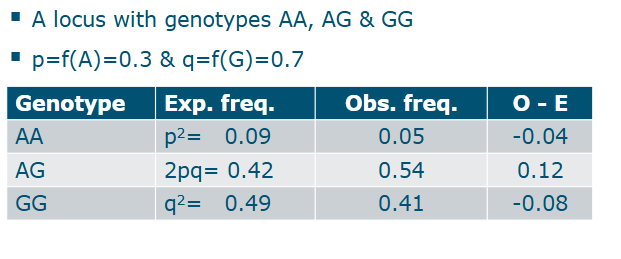
(Departures from) Hardy-Weinberg equilibrium
Sometimes loci that are not in H-W are removed
Useful to remove awkward SNPS:
Only 2 out of 3 genotyeps are observed
Only heterozygote individuals are observed
A liberal H-W threshold would remove those
But this is a bit tricky → Why could this be
Inbred populations, no random matings
Comparison of genotype to pedigree data
(Mendelian Inconsistencies)
Two main (complementary) approaches
Genomic vs pedigree relationships
Mendelian inconsistencies
Genomic vs pedigree information
Genomic informaiton may replace pedigree information in selection
Is there any use for pedigree information?
Can be used for prediction (i.e. single-step BLUP)
And for quality control
Pedigree yields expected relationships
Full sib relationship are always 0.5 (or greater, with inbreeding)
Genomic information yields actual relationships
Full sib relationship vary around 0.5
Genomic relationships capture more variance
Can be simply visualized by plotting
How to clean the data
Detect discrepancies between pedigree and genomic data
Mendelian inconsistencies
Duplication of samples (top left)
Monozygotic twins of duplication of samples (top middle)
Sample swaps or pedigree error (of full-sin or parent/offspring) right bottom
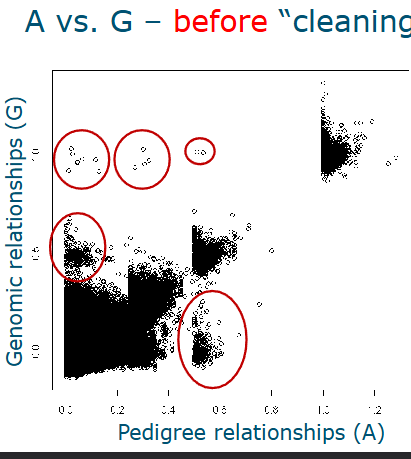
Mendelian inconsistencies (MI)
Identify animals with conflicting pedigree and SNP info
Identified by counting loci with opposing homozygotes between 2 animals
Identification is straightforward for parent-offspring pairs
Expected number of loci with opposing homozygotes = 0
MI - parent-offspring pairs
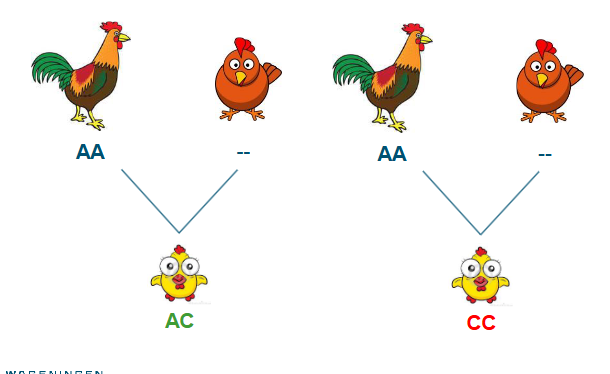
MI - paternal half sibs (dams unknown)
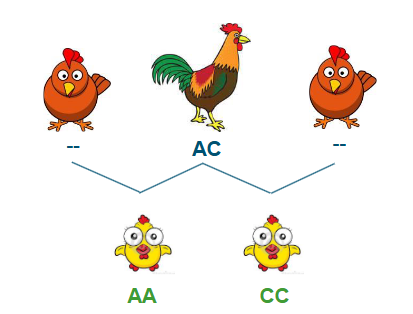
If we do not know the sires genotype (and dams)
Determine threshold # opposing homozygotes
Observed (histogram)
Expected (allele frequencies) (red)
Thresholds (empirical) (green)
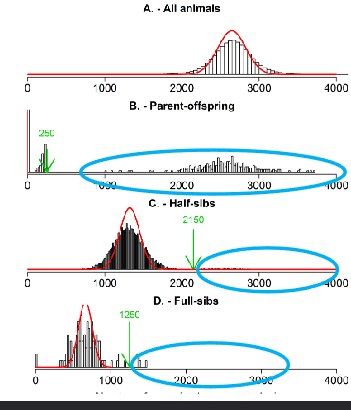
Possible causes of errors in the data
Wrong animal ID
Wrong parent ID(s)
Mistake in registration
Swapping straws of semen
Mislabelling/mixing up of DNA samples
When taking DNA sample
In genotyping process (i.e. swapping batches)
Algorithm to clean the data
Detect Mendelian Inconsistencies (starting: parent-offspring)
Detect which animal causes most inconsistencies
Pedigree or genotype error?
Remove one of the following for this animal
Genotypes
Pedigree information
Genotypes + pedigree
Repeat 1-3 untill all consistencies are removed
Evaluate A vs G relationships after cleaning
Use Mendelian inconsistencies to remove SNPs
Mendelian inconsistencies can also be used as quality control for genotype calling of individual SNPs
SNPs that show many inconsistencies in otherwise consistent parent-offspring pairs should be removed
i.e. remove SNP with > 2% inconsistencies between parent and offspring
More powerful methods
SNP based tests are sufficient for close relationships
Parent-offspring
More precise tests are required for more distant relationships
Grandparent-grandoffspring
Greatgrandparent-greatgrandoffspring
Based on counting long shared haplotypes
Long-shared haplotypes
Alternative when there is increasing distance between relatives
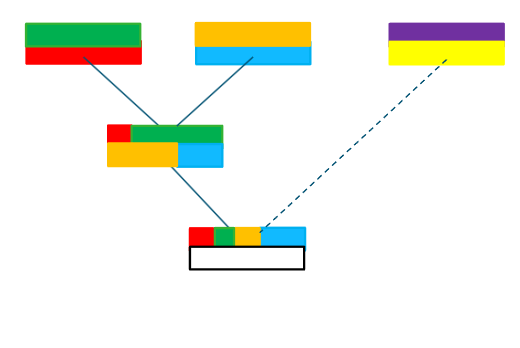
Mendelian inconsistencies (by counting opposing homozygotes) provide a …
straightforward method for quality control of genotype data
Power of the method decreases rapidly with
increasing distance between relatives
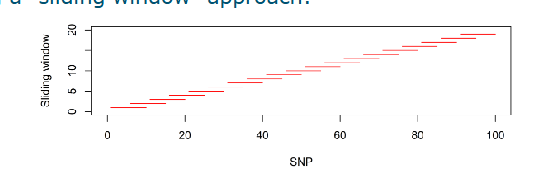
Pruning for (near) complete LD
Adjacent loci may have high LD (i.e. r² values close to 1.0)
How to remove this redundancy?
Approach
Determine an r² threshold; usually 0.95 or 0.99
Remove one SNP from adjacent pairs exceeding the threshold
Using a sliding window approach (see screenshot)
Dealing with (left-over) missing genotypes
Due to
No genotype call
Genotype removed due to mendelian inconsisteny
How to deal with those in subsequent analyses
Randomly assign based on gentoype frequencies :(
Assign the mean genotype :/
Impute using family or linkage disequilibrium (LD) info :)
Imputation using
Family information (parent-offspring, or further)
Linkage disequilibrium information
Both
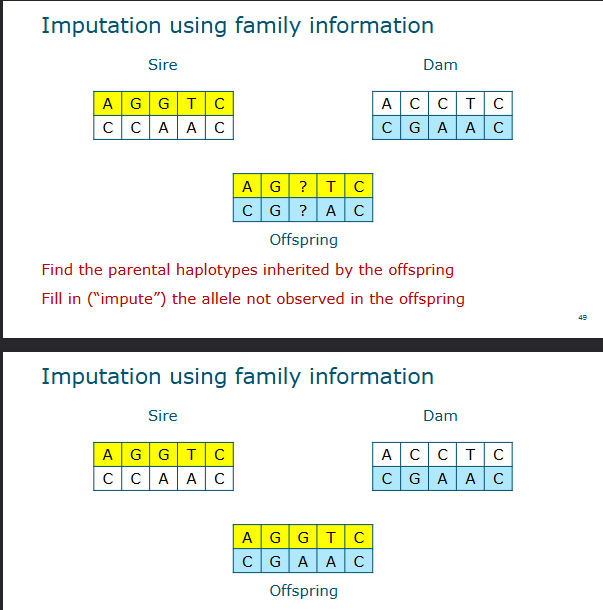
Imputation using family information
Find the parental haplotypes inherited by the offspring
Fill in (impute) the allele not observed in the offspring
Imputation using LD information
Principle the same as using family information
Now compare to all haplotypes observed in the population
Impute best mathc, or matching haplotype with highest frequency