BS3054: TOPIC THREE
1/95
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
96 Terms
Secondary Messengers
Intermediate chemicals that help transduce a chemical signal
- e.g. cAMP, cGMP, inositol triphosphate, Ca2+, diacylglycerol
What is cAMP
- second messenger
- product of reaction catalysed by adenylyl cyclase
- ATP (+ adenylyl cylase) --> cAMP
Adenylyl cyclases
- 10 isoforms
- M1 and M2 membrane bound domains
- C1 and C2 domains in the cytosol
- ATP binding site sits in-between C1 and C2 domain
- Mg2+ binding site = Mg2+ is a cofactor
- activity dependent on the Galpha subunit it receives signals from
How do isoforms of adenylyl cyclase cause specifictiy
- different isoforms respond differently to signals from G-proteins
- allows tissue specificity - not all tissues will respond to a signal and some will respond stronger than others
effectors of cAMP mediated signalling
- Protein Kinase A
- cyclic nucleotide gated channels
- cyclic nucleotide regulated GEFs
What is PKA
- protein kinase A
- serine-threonine kinase
- phosphorylates proteins
- modulated by cAMP
Structure of PKA
- two regulatory subunits
- two catalytic subunits
- 4 isoforms of R subunit (RI-alpha, RI-beta, RII-alpha, RII-beta)
- 3 isoforms of C subunit (C-alpha,beta,gamma)
w
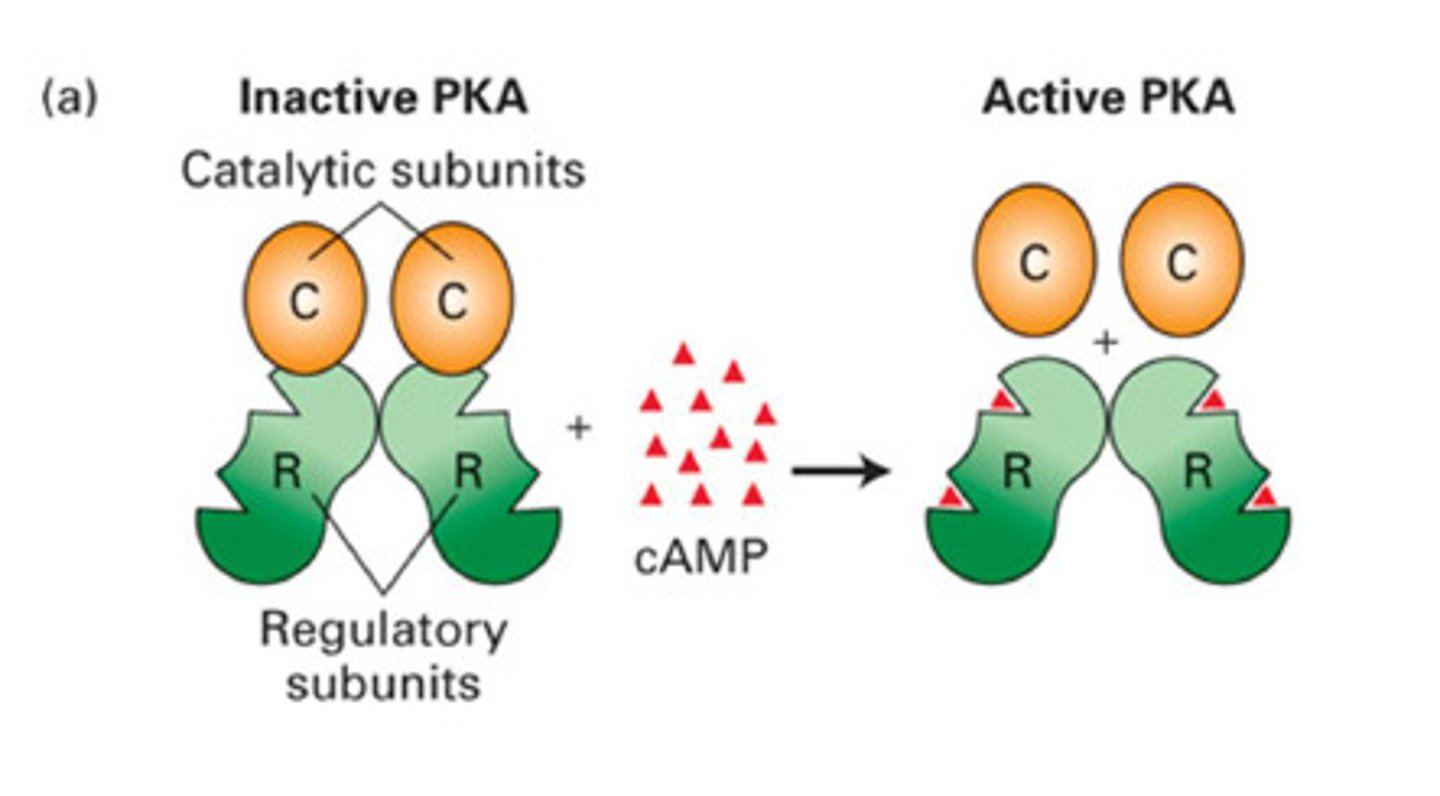
What happens when cAMP binds PKA
- cAMP binds to regulatory subunit dimer = releases the catalytic subunits from the complex
- the catalytic subunits are now active and can phosphorylate
What are the two classes of PKA
PKAI
PKAII
PKA I vs PKA II
PKA I - cytosolic
- cAMP binds to PKA and causes dissociation of the catalytic subunit = can phosphorylate substrate
PKA II - membrane bound
- docked to AKAP
- substrate binds to AKAP and then cAMP causes release of the R subunits = allows substrate to be phosphorylated
- AKAP localises PKA to specific cellular targets
What is AKAP
- A-kinase anchoring protein
Examples of PKA targets
- GPCRs, ion channels, cytoskeletal proteins, protein phosphatase and kinase inhibitors, transcription factors
Phosphodiesterase
enzyme that degrades cAMP, producing 5'AMP, to terminate signalling
- breaks phosphodiester bond
- regulated by kinases
Are there only one type of PDE (phosphodiesterase)
- 11 different families of PDEs in mammals
- some hydrolyse both cGMP and cAMP
- some preferentially hydrloyse cAMP or cGMP
guanylyl cyclases
- guanylyl cyclase catalyses GTP to cGMP
- PDE will convert cGMP to 5'GMP
What are the two types of guanylyl cyclase
1. particulate GCs = membrane bound
2. soluble GCs = in cytosol/ NO sensitive
particulate guanylyl cyclases
- transmembrane, ligand-activated homodimers
soluble guanylyl cyclases
- activated by NO and CO
- NO is an extremely potent activator
- nitric oxide binds haem group in guanylyl cyclase heterodimer
Nitric oxide
- vasodilator
- gaseous compound - only stable for seconds so made as and when needed
- detectable at very small amounts
NOS
- nitric oxide synthase
- synthesises NO
types of NOS
nNOS = neuronal + skeletal muscle = communication
iNOS = inducible = produces high NO concentrations that can exhibit direct toxic effects = immune defence
eNOS = endothelial = vasodilation
biosynthesis of Nitric oxide by NOS
- L-arginine is turned into L- citrulline and nitric oxide by NOS
Roles of NO
- activates soluble guanylyl cyclases
- nitrosylation of proteins
- direct toxicity (NO is a free radical)
major targets of cGMP
- cyclic nucleotide gated channels
- modulation of PDE activity
- activation of PKG
what is PKG
cGMP dependent protein kinase
PKG monomer
Regulatory domain
- Leucine zipper (pseudo-substrate)
- Nucleotide binding sites
Catalytic domain
- ATP site
- Kinase
= exist as a soluble homodimer
what is a pseudo-substrate
- substance that mimics the real substrate of an enzyme
- often part of enzyme own structure
- blocks enzyme active site and inhibits activity
How does NO cause vasodilation
- NO activates soluble guanylyl cyclase
- increases levels of cGMP within smooth muscle cells of blood vessel walls
- rise in cGMP leads to activation of PKG
- PKG activates SERCA pump to move calcium ions from cytoplasms to ER
- cGMP can activate potassium channels causing hyperpolarisation = closes VGCC = promoting relaxation
Phospholipid structure
2 fatty acids, 1 glycerol, 1 phosphate group
- hydrophilic head and hydrophobic tail = amphipathic
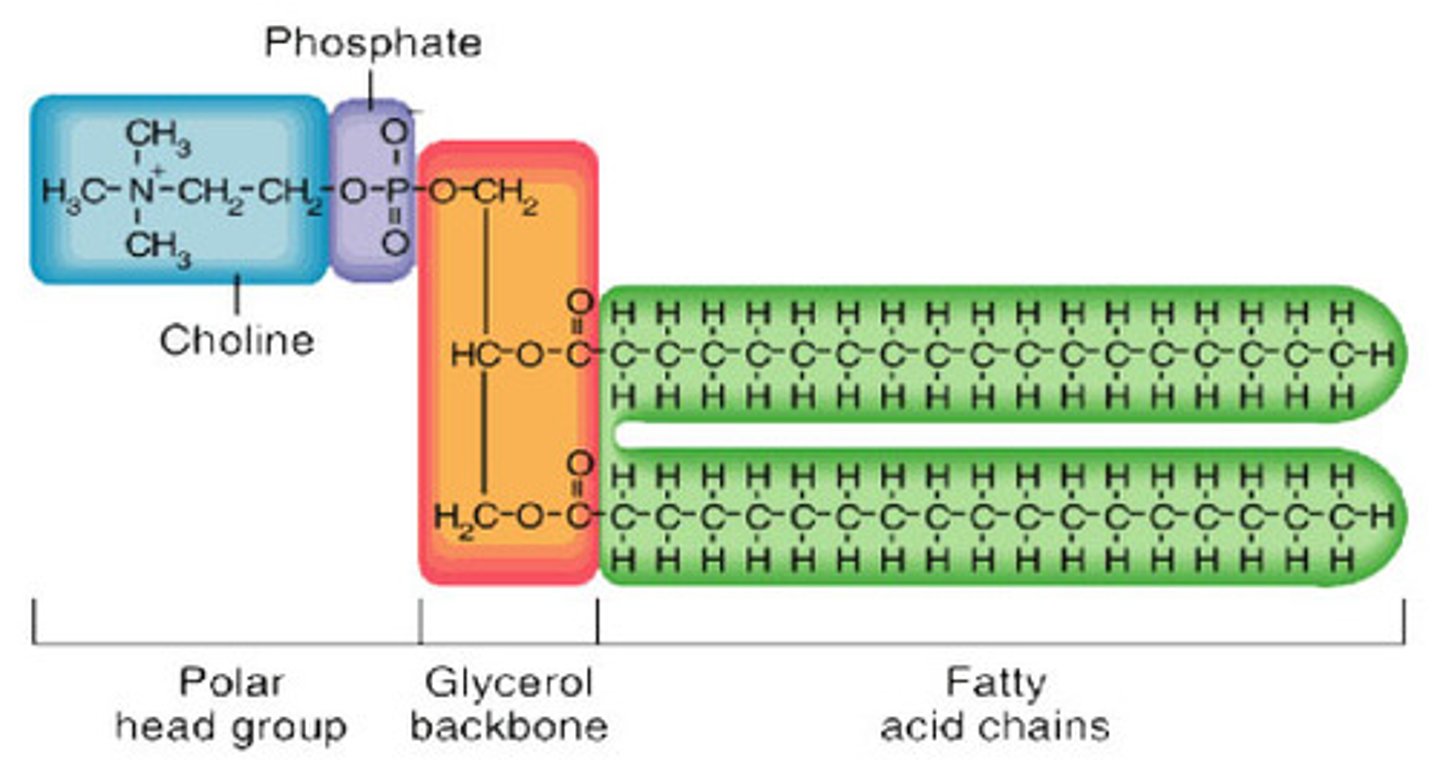
X group on phosphate of phospholipids
- can add groups
e.g. add choline to make phosphatidylcholine
e.g. add inositol to make phosphatidylinositol
phospholipids classified according to their polar head group and their abundance
- phosphatidylcholine = 50% of membrane lipids
- phosphatidylserine = 2-10%
- phosphatidylethanolamine = 15-35%
- phosphatidylinositol = 5-10%
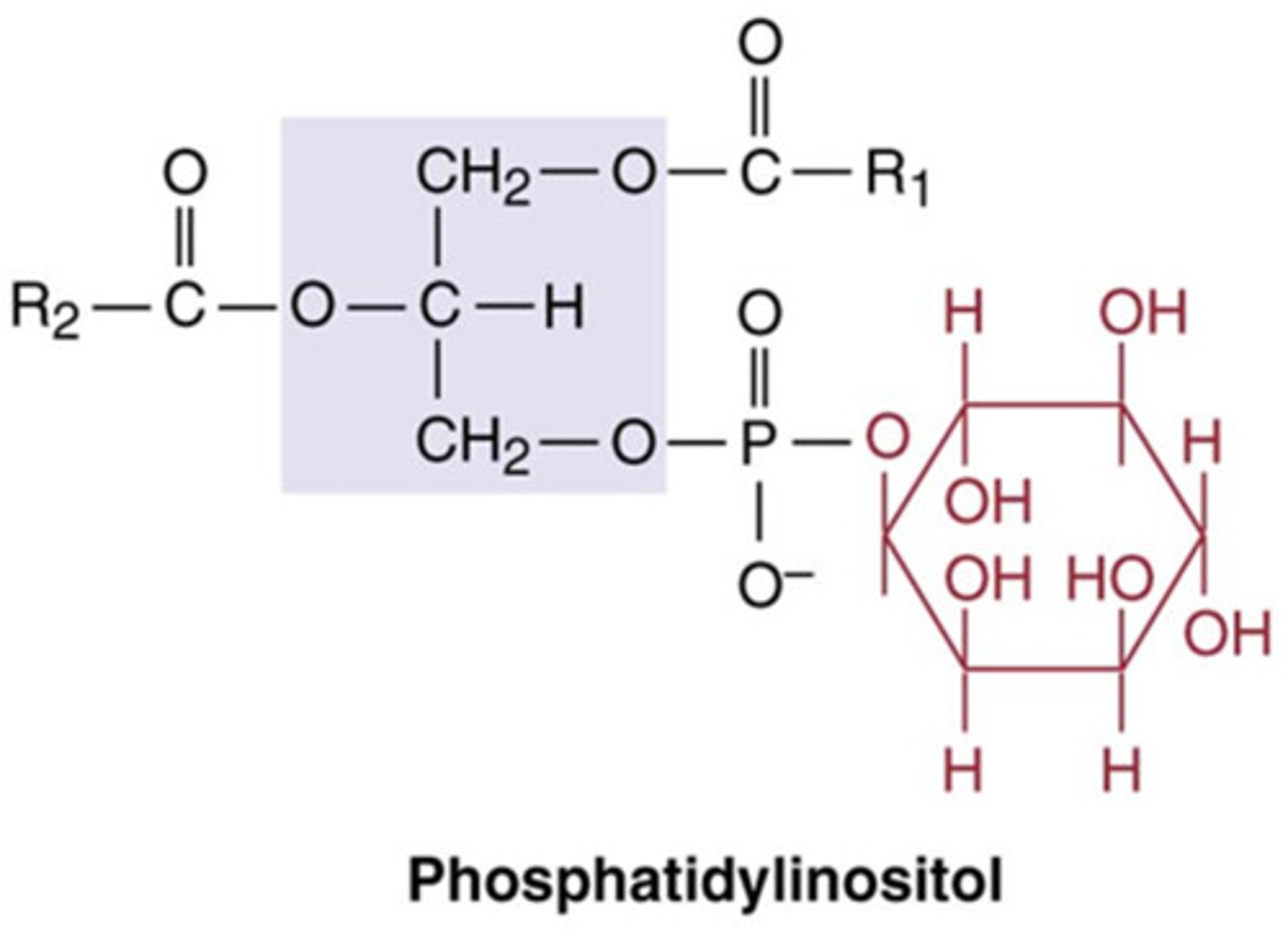
Other denotations of phosphatidylinositol
PI or PtdIns
How is PI modified
- phosphorylation of any of the 6 carbons on the inositol group make other signalling molecules e.g. PIP2
Variations of PI
- phosphatidylinositol = PI
- phosphatidylinositol 4-phosphate = PIP
- phosphatidylinositol (4,5) - bisphosphate = PIP2
- phosphatidylinositol (3,4,5) - triphosphate = PIP3
why is PIP2 called phosphatidylinositol 4,5-bisphosphate not called phosphatidylinositol 4,5-diphosphate
- because the phosphates are on different carbons they are not organised in a chain where it would be referred to as diphosphate
Which carbons is PIP2 phosphorylated on
carbons 4 and 5
phosphatidylinositol structure
what enzymes turn phospholipids into signalling second messenger molecules
phospholipases
variations of phospholipases
- PLA1
- PLA2
- PLD
- PLC
= same substrate different outcomes
Role of phospholipase C
- cleaves PIP2 between the oxygen on the glycerol backbone and the phosphate
- forms DAG and IP3 = both second messengers
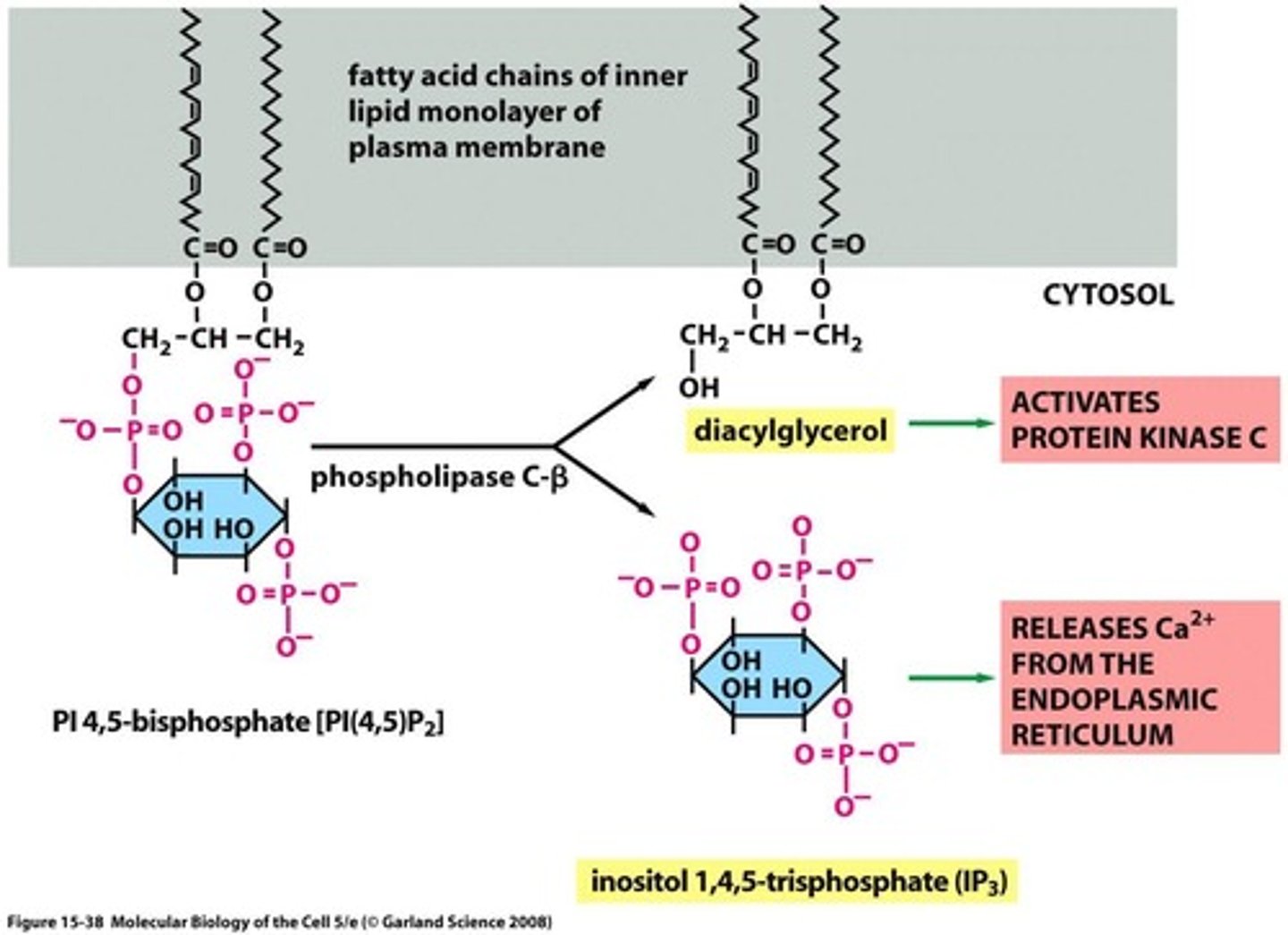
what is IP3
inositol 1,4,5-triphosphate
Receptor mediated signalling through PLC
- agonist binds receptor = conformational change
- alpha subinit dissociation and GDP/GTP exchange
- recruitment of PLC = cleaves PIP2 = DAG + IP3
- IP3 stimulates release of calcium from IC stores (ER) by binding to IP3 receptor
- DAG and calcium activates PKC
IP3 receptor
- acts as coincidence detector
- requires both IP3 to bind and calcium to be present for ion channel to open and cause calcium release into cytoplasm = calcium induced calcium release
families and isoforms of PLC
6 families, 13 isoforms (30 splice variants):
- PLC-beta (4 isoforms)
- PLC - gamma (2 isoforms)
- PLC - delta (3 isoforms)
- PLC - epsilon (1 isoform)
- PLC - zeta (1 isoform)
- pLC - eta (2 isoforms)
why is it important for there to be different isoforms of PLC
- to allow precise control so signalling can be regualted between cell types/locations
PLC-beta
- has 4 isoforms PLC-b1-4
- X and Y catalytic domains
- PH domain = allows localisation = bind PIs
- 4 tandem EF-hand domains = calcium function but unclear
- C2 domain = Ca2+ binding
- CC domain and PDZ domain = protein-protein interactions with PLC
homology between isoforms of PLC-beta
- 60% homology of the catalytic domain between isoforms
what is PLC-b most commonly associated with regulating
GPCRs
PLC-beta isoforms tissue distribution
B1 and B3 = fairly widespread
B2 = immune/haematopoietic
B4 = retina and certain neurons
What can activate PLC-beta
- Gaq subunit
- G beta-gamma subunit
- Ca2+
what domains does G-beta-gamma subunit associate with on PLC-beta
- PH domain (localisation of PIs)
- catalytic region
- almost always beta-gamma subunit from Gai/10 proteins as they are the most abundant
PLC-beta role other than phospholipase activity
- GTPase activating proteins
- can bind to alpha subunit and speed up intrinsic GTPase activity to cause reassociation between alpha and beta-gamma subunit
- same role as RGS proteins
coincidence detection with G-proteins and PLC-B3
- Gaq and Gbeta-gamma both activate PLC-B3 = synergistic activation
how is inositol recycled
- dephosphorylation of IP3 creates inositol
- inositiol fed back into membrane where it is phosphorylated into PIP2 again by kinases
- PIP2 can be turned into IP3 and DAG again or phosphorylated by phosphoinositide 3-kinase into PIP3 = signalling
phosphoinositide kianses
- phosphorylate PIs at 3,4,5 positions on inositol
- phosphatases dephosphorylate these
e.g. phosphoinositide 3-kinase phosphorylates in the 3 carbon position
PI3Ks
= phosphatidylinositol 3-kinases
= phosphorylate in the 3-OH position of the inositol ring in PIs
- 3 main classes = I, II, III
- activated by diverse cell surface receptors mainly RTKs and some GPCRs through Src transactivation of RTKs
- its preferred substrate in vivo is PIP2 = converts it to PIP3
GPCR singalling via Src transactivation of RTKs
- agonist binding to GPCRs can cause Src activation
- Src can then phosphorylate and activate RTKs
- transactivation amplifies the signal by integrating different pathways onto the RTK cascade
Structure and function of PI3-kinase
Regulatory subunit:
- p85 with SH2 and SH3 domains associated = allows recruitment to RTKs or adaptor proteins
Catalytic subunit
- p85 binding domain
- Ras binding domain - Ras binding can activate catalytic subunit
- HR3 = membrane binding
- HR2 = scaffold for other proteins to bind to it
- HR1 = kinase core
Why is PI3k associated with cancer
- Ras binding to PI3k causes cell proliferation
activation of PI3 kinase via RTKs
- growth factor binds to RTK causinf autophosphorylation and dimerisation
- allows docking of Grb2, SOS, RAS and GTP
- PI13k docks via Ras domain
= production of PIP3 on membrane
PIP3 as an anchor for signalling proteins
- signalling proteins with PH domains accumulate at sites of PI3K activation by binding to PIP3
- these proteins regulate cell growth, survival and movement
- examples of proteins containing PH domains are PKB (Akt) and PDK1
PKB (Akt)
- serine/threonine kinase
- growth factor pathway
- activated by PI3K and PDK1/2
PDK1
phosphoinositide dependent kinase 1 (involved in activation of PKB)
PKB activation via PDK1
- PIP3 in the membrane recruits PKB (Akt) and PDK1
- PDK1 phosphorylates PKB to partially activate it
- mTORC2 complex further phosphorylates AKT to fully activate it
- activated AKT then inhibits the TSC complex leading to activation of mTORC1 = controls protein synthesis and growth
What is PKB/Akt generally associated with
anti-apoptosis, growth, proliferation and migration
Termination of PI3-kinase signalling
- SHIP proteins remove binding sites for proteins with PIP selective PH domain
- SHIP proteins generate PIP2 from PIP3 that PKB can bind to
- PH domain of PKB binds PIP2 and PIP3 with equal affinity
- PTEN turns PIP3 into PIP2 and PIP2 into PI
PI3-kinase signalling and cancer
- numerous oncogenes activate type IA PI3-kinase
- activating mutation of PI3-kinase described in cancer
- PTEN has tumour supressor properties - mutations in PTEN associated with cancer
- mutations in SHIP1 recently associated with some leukaemias
therapeutic attempts to inhibit PI3-kinase in cancers
- small non-specific molecules
- wortmannin
- LY294002
- copanlisib and apelisib - only active on one kinase (class I PI1K inhibitors)
- most approved drugs have since been withdrawn due to side effects
Theoretical ideal therapeutic targets for PI3K in cancer
Isotype selective PI3K inhibitors:
- inhibitors that target specific p110 catalytic subunits - many minimise side effects
Inhibitors of Akt (not yet apporoved)
- inhibition of downstream signalling from PI3K activation many be beneficial
- two examples of Akt inhibitors:
1. ipatasertib = binds ATP binding site of Akt (breast cancer)
2. afuresertib = competitive inhibitor
Why is calcium important to the body?
- strong bones and teeth
- fertilisation
- proliferation
- neurotransmission
- apoptosis
- contraction
- gene expression
Intracellular vs extracellular calcium conc
- Calcium concentration is much higher outside the cell than in the cell cytoplasm
- 1-2mM (10^-3) outside
- 100nM (10^-7) inside
What is the equation for the driving force of calcium movment
Nernst equation
E = RT/zF ln ([ion outside] / [ion inside])
Advantages of IC calcium conc being much lower than EC
- this is good because it means only a small amount of calcium needs to move into the cell in order to create a large change
- the balance can also be restored easily if not much calcium has to move
Disadvantages of IC calcium conc being much lower than EC
- to remove ions from the cell requires a lot of ATP
- cell can become overloaded with calcium easily which leads to cell death
How is the calcium gradient set up and maintained
1. impermeability of membrane
2. Ca2+ release across plasma membrane
- Ca2+-ATPase
- Na/Ca exchanger
3. Ca2+ buffers
4. Intracellular Ca2+ stores
1. Impermeability of membrane in calcium gradient set up
- hydrophobic lipids in the membrane = relatively impermeable
- once calcium has left cell it is hard for it to get back in
= maintains higher conc outside than in cell
2. Ca2+ release across plasma membrane in calcium gradient set up
Ca2+ ATPase (PMCA) - plasma membrane calcium atpase
- sits in plasma membrane
- drives calcium out of cell using ATP
- high affinity so can bind even with low levels of calcium inside cell
- low capacity = saturates quickly so not very efficient
Na+/Ca exchanger
- electrochemical gradient of Na+ means that Na+ moves from outside of cells to inside
- 3 Na+ move into the cell and take 1 Ca2+ with them
= antiport system
- this system is electrogeneic = works best at resting membrane potential
- low affinity but high capacity
Ca2+-ATPase pump (PMCA)
- calcium conc increases in the cell
- calcium binds to calmodulin and activates it
- calmodulin affinity for calcium increases upon calcium binding
- caclium-calmodulin binds to Ca2+-ATPase
- Ca2+-ATPase removes calcium from the cytoplasm into the EC space
3. Calcium buffers in calcium gradient set up
- when calcium enters the cell it doesn't travel far
- it gets tied up in calcium binding proteins e.g. parvalbumin, calreticulin
- calcium will only travel through if these proteins are saturated when there is a large calcium influx
- regulates the amount of calcium needed to initiate a cellular response
4. Intracellular stores of calcium in gradient set up
- calcium is stored in the SR/ER of the cell
- IP3 binding to receptors or Ca2+ binding to ryanodine receptors (calcium induced calcium release) = causes calcium release from ER into cytoplasm
- SERCA pump reuptakes calcium into SR/ER
How are calcium levels elevated and returned to basal levels
1. influx across plasma membrane
- voltage-gated Ca2+ channels
- inotropic receptors = ligand-gated calcium channels
2. rapid release from EC/SR
3. Golgi, nucleus and mitochondria (non-rapid) can also serve as Ca2+ stores
How is ca2+ influx across the membrane regulated
VOCCs
- channels sense membrane depolarisation and change shape = opens
- negative amino acid residues on the channel attract positive calcium = calcium moved through the channel into the cell along conc gradient
Ligand-gated ion channels (inotropic)
- ligand binds receptor and causes channel to open
- calcium will flow into the cell along concentration gradient
e.g. ACh binding receptors allowing Ca2+ influx
How is Ca2+ release from IC stores regulated
Release into cytoplasm from ER:
- GPCR activation
- Activation of PLC
- PIP2 -> DAG and IP3
- IP3 binds receptors on ER and stimulates Ca2+ release into cytoplasm
= ligand-gated ion channel but on ER membrane not plasma membrane
- Ca2+ now in the cytoplasm opens ryanodine receptors on ER membrane which causes further release of calcium from the ER = calcium induced calcium release
Re-uptake into the ER = SERCA pump
- Ca2+-ATPase that pumps Ca2+ from the cytoplasm into the ER/SR
IP3 and ryanodine receptors on SR/ER
- regulated by calcium
- when calcium is low it stimulates them to open
- at high calcium conc in cytoplasm ryanodine and IP3 receptors are inhibited
Why are there calcium binding proteins inside ER
- it calcium reacts with phosphates it forms a solid
- binding proteins allow high levels of Ca in ER without it solididying
IP3 receptors
- tetramer
- 4 subunits
- ligand gated ca2+ channel
- IP3 binds outside
Why is calcium stored in nucleus
- the nuclear envelope is continuous with the ER
Non-rapid releasable store of calcium - mitochondria
- when calcium levels are very high mitochondria will take up calcium to protect the cell from death
- microdomains = if calcium flood into the cell close to the mitochondria then that site will be highly concentrated = localised high concentration of calcium
= the mitochondria will take up the calcium
- mitochondria dont release calcium easily
What is mitochondrial calcium uptake driven by
- large negative inner membrane potential
- voltage dependent anion channels
- ca2+ uniporters of inner membrane
Why do mitochondria take up calcium
- calcium buffering
= regulate calcium levels if in excess - can release later and more slowly
- stimulation of mitochondrial metaboism
= calcium overload in cells requires mito to produce more ATP to power pumps to remove calcium
- cell death
= avoids cell death from overload of calcium
small calcium stores
lysosomes, endosomes, phagosomes, secretory vesicles
Store refilling and restoration of calcium concentration
- too much calcium for too long is toxic so levels have to return to basal
= termination of signal (desensitisation or ligand dissociation/reuptake)
= calcium removal
= Ca2+ store refilling
How are calcium stores refilled
- recycling of cytosolic calcium that was released from ER
- capacitative Ca2+ entry
Capacitative Ca2+ entry
- capacitative or store-operated channel (SOC)
- system that tells the cell that the ER stores are depleted
- SOC sits in the membrane and interacts with the SERCA pump
- STIM1 sits in ER membrane = detects ER stores are low and binds calcium = activation of STIM1
- STIM1 aggregates together and localises by the CRAC channels = CC domains on CRAC channels and STIM1 interact
- = CRAC channels open = allow calcium back into the ER
Synaptotagmin
- calcium sensor that affects NT vesicle release
- synaptotagmin sits in membrane of vesicle and binds calcium - when bound to calcium it has higher affinity for syntaxin
- C2A and C2B domains interact with syntaxin on the plasma membrane
- allows fusion of vesicles to the membrane for NT release
How does calcium regulate so many different processes
SPACE
- dependent on location of influx - from stores or plasma membrane
TIME
- long exposure to calcium avoided by low amplitude signals to make calcium release slow and transient
- depending on the receptors and the tissue types, calcium release and uptake is different
AMPLITUDE
- strength of signal impacts response