chem - exam
1/23
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
24 Terms
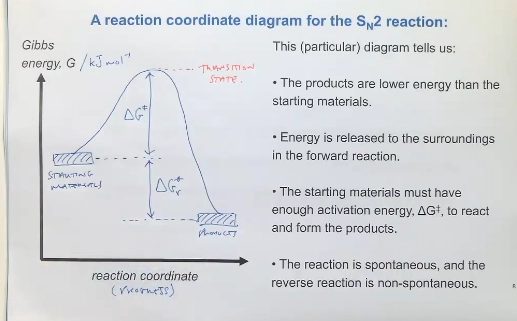
list the aspects of a Reaction Coordinate diagram
X axis (Gibbs energy, G/kJ mol-1)
Y axis (Reaction progress)
energy of reactants & products (specify the structure of these), and energy between these (DeltaGr)
energy barrier deltaG* (to pass through the transition state, and reach the products) - energy maxima
intermediate state (species in between multiple elementary steps)
how are these related to an RC diagram (and rxns in general)
thermodynamics
kinetics
(thermodynamics)
if a reaction is spontaneous to occur by itself
products with lower enery, are more stable, so are spontaneous (energy releaesd, deltaGr negative) - relative stability of reactants & products
position of equilibrium
(kinetics)
how fast a reaction will occur, if the reactants have enough Ea to overcome the E barrier and form the products
the size of the E barrier (& so considers catalysis to lower E barrier)
movement of e & how bonds break and form (Rxn mechanisms!), using the rate data (how slow and fast these are, RDS)
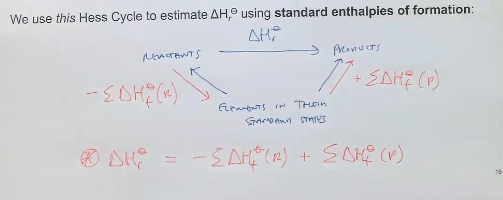
what are hess’s cycles, what do these help us calculate
estimating deltarH (enthalpy change) for reactions at standard states
(Standard enthalpy of formation)
thinking about elements at standard states (elemental states), the deltarHs to form these - which are all tabulated
combining these, flipping equations, manipulating stoichometries - we can find the deltarHs of reactants and products
(bond dissociation energies)
looking at the enthalpy required to break 1 mole of a given bond in the gas phase, to form gaseous individual atoms (usually as radicals, with one e from the bond going to each species)
these energies are always positive (energy absorbed to break bonds, energy released when forming bonds)
we can then think of the bonds broken and formed in reactants → products of our reaction, use the tabulated data to see their enthalpies
then combining with the equatio n (detlarH = sum of bonds broken - sum of bonds formed)
what is spontaneity?
with what parameter is this measured
what aspect (TD vs K) is related
what law underpins this
(spontaneity)
spontaneous reactions once started, will continue without outside intervention (ball rolling down the hill)
isnt just told by exo or endothermic (requires a consideration of entropy, enthalpy change, and T)
doesnt tell us about the rxn speed (thus is a THERMODYNAMIC concept)
(entropy)
denoted S, a measure of randomness, deltaS saying the difference in disorder between reactants and products
positive deltaS (less order, disorder created)
negative deltaS (more order, order created)
delta S 0 (at equilibrium)
(2nd law)
the net entropy (disorder) of the universe tends to increase, each transfer in energy increases disorder of the universe
even with local increases in order (e.g. protein folding, cellular processes), there is net decrease in order (increase in entropy) of the surroundings
(kinetics)
spontaneous rxns, can be SUPER SLOW (not realistic in life), as this doesnt consider kinetics (Rxn speed)
what equation (and main parameter) is used to determine spontenaity
Gibbs free energy! deltaG
negative deltaG (spontaneous)
positive deltaG (non spontaneous)
zero deltaG (at equilibrium)
(equation)
deltaG = deltaH - T * deltaS
tells us if a rxn is spontaneous, considering T, deltaH (Exo vs endo), and deltaS (change in order)
tells us if products are more stable than reactants, accounting for both enthalpy & entropy
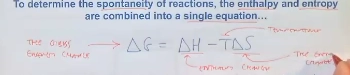
how do each of the components of the deltaG equation, affect deltaG (rxn spontaneity)
positive deltaG (non spon), negative deltaG (spon)
sign of deltaG, depends on how negative or positive, deltaH is compared to T*deltaS
however, T has quite a large effect - most of the time we can make a rxn go just by changing the T
(TEMPERATURE INDEPENDENT)
(-deltaG) +deltaS (disorder) & -deltaH (exo, release E), will always be spontaneous
(+deltaG) -deltaS (order) & +deltaH (endo, absorb E), will always be nonspontaneous
(TEMPERATURE DEPENDENT)
-deltaS (order) - deltaH (exo, release E)
will be spontaneous at low T (T*deltaS contribution will be small to -deltaH, so deltaG remains negative)
will be nonspontaneous at high T (T+deltaS will have large positive contribution to -deltaH, so deltaG becomes positive)
+detlaS (disorder), +deltaH (endo, absorb E)
will be spontnaoeus at high T (T*deltaS will have a large negative contribution to +deltaH, so deltaG becomes negative)
will be nonspontaneous at low T (T*deltaS will have a small negative contribution to +deltaH, so deltaG stays positive)
what is an extreme ecample of T dependence in reaction spontaneity
(polymerisation!)
taking many monomers (weak pi bonds), and making one big molecule (strong sigma bonds)
thus having a large release of energy (low deltaH), but a large increase in order (low deltaS)
so T largely becomes the decider in deltaG (spontaneity) as the rxn proceeds, and can control polymer chain length
this is because, as T increases, -T*deltaS becomes larger than the large -deltaH, so deltaG becomes positive, and the reaction ceases to proceed spontaneously
how does deltaG relate to K (Eq constant)
how can they be used together to tell us about a rxn
(K)
tells us the composition of a mixture at dynamic eq (equal rate of forwards & reverse rxns), the conc. ratio of products / reactants
is TD, so doesnt tell us about the rxn speeds
large K = more products (gone to completion)
zero K = equal
negative K = more reactants
(relation to deltaG)
deltaG = -RT lnK
K = e^ -deltaG / RT
due to this exponential factor (e), we see large changes in K with small changes in deltaG
(their use together)
looking at K & deltaG together, tells us about rxn mixture composition at eq
(completion) large K, low deltaG (spontaneous, released energy)
(no rxn) low K, large deltaG (nonspon)
(reversible process) intermediate K (10^-3 ←> 10³), intermediate deltaG (-22.8kj <=> 22.8kJ)
how can eq’s position be affected
what principle underpins this
why is this important for biochemists
controlling rxn conditions, influences the position of eq by influencing K (Ratio of products/reactants, what is produced more)
can change temp, conc - and eq will respond by changing position (changing what it favors) to minimise the change (Le CHateilier’s Principle)
important for biochemists, to bias rxn towards the product we want to synthesise
how can T affect K (position of eq)
what equation underpins this
exothermic is one direction (RELEASES HEAT), endothermic is one direction (ABSORBS HEAT)
(exothermic)
lowering T favors products, as this rxn releases heat to minimise the change (K increases)
increasing T favors reactants, as they want less heat released, they want to absorb the added heat, so endothermic reverse rxn is favored (K decreases)
(endothermic)
increasing T favors products, as this rxn absorbs the added heat (K increases)
decreasing T favors reactants, as this absorbs less heat, they want to actually warm up the environment, so the reverse is favored (K decreases)
(Van’t Hoff equation)
dont need to rememebr, but just know that K is inversely proportionall to - deltaH / RT (as -deltaH / RT increases, K decreases)
(endothermic) as T increaes, this contribution becomes more negative, so K becomes larger
(exothermic) as T increases, this contribution becomes more positive, so K becomes smaller

how can reactant & product conc. affect K
why is this important for chemists
K = conc products / conc reactants
therefpre changing conc. of these components, directly affects rxn outcome to favor a certain direction (stoichometry)
(incr reactants) acts to favor the forwards rxn to use up the added reactants and make more products (K increases)
(incr products) acts to favor the reverse, to use up added products and make more reactants (K decreases)
(use in chemistry)
to create the product we want, we can add an excess of the reactant, for those that are cheaper / readily available, to ensure more forwards rxn occurs
even if the other reactant isnt increased, it still means that it has a higher chance of reacting more, with more of the other reactant around
can also remove side products as they form, to favor the forwards reaction, to minimise the change by creating more products (and because theres less material for the reverse rxn to occur)
even if not removing our desired product, we still are removing a product, and favoring forwards rxn
e.g. evaporating water via distillation, removing things like water via molecular sieves
how does protein folding happen and abide by the 2nd law, if it increases order?
it increases order locally (protein is folded into a more stable structure), but disorder (entropy) increases overall, as water is released from the protein & solvent into the surrounds (incr disorder, more molecuels are formed from the starting materials)
rationalise Reaction Coordinate diagrams in terms of the kinetics underpinning them
RC diagrams describe HOW reactions occur
reactants must collide with enough energy (meeting or exceeding Ea deltaG*), to break the bonds in reactants and form bonds in the products
each elementary step (at least one per rxn, can have multiple in a multistep rxn) has this Ea to overcome
as they reach the top of the Ea barrier, they pass through a TS, which is the structure in between breaking and forming bonds
TS have no lifetime (not long enough to isolate / observe / track), but we know their energy is the highest part of the E profile
what is the difference between…
rxns with 1 elementary step
rxns with multiple elementary steps
(one elementary step)
for exothermic (examples disccussed in this context)
reactants are higher energy than products, but to form products must pass through an Ea barrier (matching deltaG*)
at the top of the E barrier being the TS (in between of breaking & forming bonds)
deltaGr for the rxn being the distance between E of reactants & products
e.g. Sn2 reaction (NuSub bimolecucalr)
(multiple elementary steps)
fundamental ideas in the elementary step, but just with multiple of these
between the TSs of subsequent elementary steps (deltaG* 1, deltaG* 2….), they form reactive intermediates (local energy minimum)
these are short lived, having higher E than materials or products, and are more observable than TSs
different elementary steps combined, will have different Ea barriers, so whichever is higher is the RDS (slower elementary step, therefore it determines the rxn rate)
e.g. Sn1 reaction (NuSub unimolecular)
what is the Hammond Postulate?
why is this useful?
along a single reaction pathway (an elementary step), the TS will most closely resemble, the species it is most energetically close to
so a TS will resemble the reactant if higher energy, or the product if higher energy
so for multistep rxns, a TS may also resemble the reactive intermediate, if energetically closest
(important?)
we can use this to predict the structure of the intermediate in a rxn
how does T relate to rxn rate?
described by the Arrhenius equation, considering the Ea, R, T, and proportion of collisions that give rxn
rate rapidly increases with increasing T, as we have more frequent collisions that give rxn