Unit 2 Orgo II Concepts
1/65
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
66 Terms
What are the four criteria for aromaticity?
Cyclic: system must be a ring
Planar: ring must be flat, allowing p-orbitals to overlap
Fully Conjugated: every atom in the ring must have an available p-orbital (no sp3 gaps)
Huckel’s Rule: the system must have 4N + 2 pi electrons (n must be an integer, set 4n+2=# of pi electrons)
What does it mean to be antiaromatic?
Meets criteria of being cyclic, planar, and fully conjugated, but has 4n pi electrons, and are exceptionally unstable.
What does it mean to be non-aromatic?
Fails any of the first three criteria of being cyclic, planar, and fully conjugated
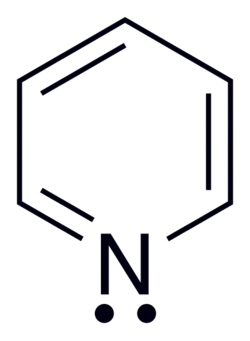
Pyridine & Aromaticity
Nitrogen lone pair is in an sp2 orbital perpendicular to the pi system. Does not count toward the 6 electrons, and is basic. It juts out and is not involved in resonance as the pi bond attached already is.
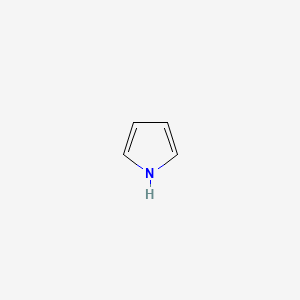
Pyrrole and Aromaticity
The nitrogen lone pair is part of the aromatic sextet to reach 6 electrons. It is not basic because using the lone pair to grab a proton would destroy aromaticity, which is highly unfavorable.
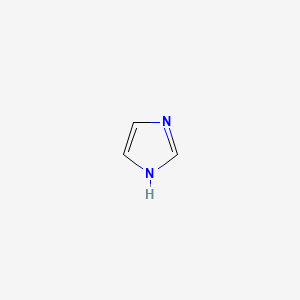
Imidazole and Aromaticity
Contains both types, so that one nitrogen is pyridine like (basic, a lone pair to give) and one is pyrrole-like (non-basic)
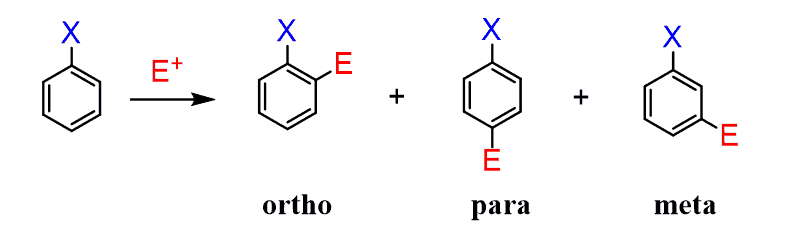
What are the disubstituted patterns?
Benzene Physical Properties: Resonance
Much more stable than expected.
Benzene Physical Properties: NMR
Because of the resonance ring current, aromatic protons are highly deshielded and appear at 7-8 ppm.
Are polycyclic aromatics more or less stable than benzenes?
Their transition state is more stabilized because the aromaticity of the second ring remains intact, while for benzene, it breaks entirely. This results in a lower loss of resonance energy compared to benzene, so it occurs faster in reacting than regular benzenes.
Polycyclic aromatics are more stable than isolated alkenes, but they are less stable (more reactive) on a per-ring basis.
Benzene treated with H2 and Pd/C at room temp
Result: No Rxn
Ignoring Theory Result: Rid double bond, add H on each side.
Why: benzene is resistant to hydrogenation compared to regular alkenes. You need extreme pressure/heat/input of energy with Nickel
Electrophilic Addition on Benzenes (Br2 or HCl)
Result: No reaction
Why: adding across double bond would destroy the aromaticity, prefers substitution as it preserves aromaticity and merely replaces an H with a halogen, etc.
Reaction of Pyridine + HCl
Acid-Base Reaction
Result: nitrogen gets protonated to form a pyridinium salt bc lone pair is donatable/basic
Reaction of Pyrrole + HCl
Acid-Base Reaction
Result: no reaction, as no basic lone pair
Formation of Aromatic Ions: non-aromatic species—> aromatic species using a base
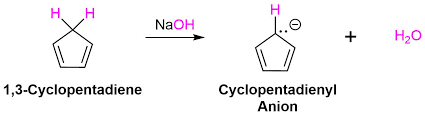
Cyclopentadiene + NaH: base pulls off a proton from the sp3 carbon to create the cyclopentadienyl anion (brings sp3 gap to aromatic, 6 pi electrons)
Electrophilic Aromatic Substitution (EAS) Overview
Reactants: Benzene (nucleophile)
Reagent: Strong catalyst/electrophile
Products: substituted aromatic ring. one hydrogen is replaced by the electrophile.
Electrophilic Aromatic Substitution (EAS) Mechanism Overview
Attack on Electrophile: benzene pi electrons attack strong electrophile (E+) creating a resonance stabilized carbocation called the Sigma Complex
Deprotonation: a base in the mixture pulls the proton off the sp3 carbon, allowing the electrons to collapse back into the ring to recreate the aromatic system
What is the sigma complex in aromatics?
Helps to predict where groups go on a ring, state when the molecule is no longer aromatic but the charge is delocalized over the remaining 5 carbons
EAS Bromination
Reactant: alkene
Reagents: Br2/FeBr3
Electrophile: Br+
Product: addition of 1 Br on molecule
EAS Chlorination
Reactant: alkene
Reagents: AlCl3, Cl2
Electrophile: Cl+
Product: alkene aromatic with 1 Cl attached
EAS Nitration
Reactant: alkene aromatic
Reagents: HNO3/H2SO4
Electrophile: NO2+
Product: Alkene aromatic with 1 NO2 group attached
EAS Sulfonation
Reactant: alkene aromatic
Reagent: SO3, H2SO4
Electrophile: SO3
Unique: reversible. add dilute acid and heat to remove the group (desulfonation)
Desulfonation
Reactant: Benzene with SO3 attached
Reagent: dilute H2SO4, heat
Product: Benzene, removal of SO3 group
Friedal-Crafts Alkylation
Reactant: Benzene
Reagent: alkyl halide (R—Cl) / AlCl3
Electrophile: R+
Product: Benzene with R group attached
Note: watch out for carbocation rearrangements. a primary alkyl halide will often rearrange to a secondary or tertiary carbocation before attacking the ring to stabilize.
Friedal-Crafts Acylation
Reactant: Benzene
Reagent: Acyl halide (RCOCl) / AlCl3
Electrophile: RC(triple bond)O+
Product: Benzene with R—C=O bond attached and HCl
Advantage: no rearrangements here, and the product is deactivated, preventing multiple additions
How many times does each EAS group add? And why?
Nitration (NO2), Chlorination (Cl), Bromination (Br), and Sulfonation (SO3H), Friedal Crafts Acylation (COR), all add 1 time because they are all deactivating. Once the first group is on the ring, it pulls electron density away, making the ring a worse nucleophile, so it cannot attack as well.
Friedal Alkylation (R) adds multiple times (polyalkylation) because the R groups are activators, so once you add one alkyl group, the ring becomes more reactive than the starting benzene due to inductive electron density donating effect. The activated ring then hunts down more electrophiles faster than the original benzene can.
When do substituent effects come into play?
When a group is already on the ring
When a group is already on the benzene ring, what does it control?
Reactivity (how fast the reaction occurs) and orientation (where the groups add)
Activator
Para/Ortho directing
Donate electron density into the ring, making it a better nucleophile
Positive charge in sigma complex stabilized directly by the lone pair of an activator when at para or ortho positions
Activator Examples
NH2, OH, OR (donate via lone pair resonance)
Moderate/Weak: alkyl groups (donate via induction)
Deactivator
Meta Directing
Pull electron density out of the ring, making it a worse nucleophile
Place positive charge or partial positive next to the ring (on the atom just next to the carbons on the ring), and attacking ortho/para would place the sigma complex’s positive charge directly next to the group’s positive charge, making it electronically repulsive, so they direct to the meta position (least bad option)
Deactivator Exception
Halogens: F, Cl, Br, I
Deactivators because they are electronegative but they are ortho/para directors because they have lone pairs that can stabilize the sigma complex via resonance
Deactivator Examples
NO2, CN, CHO, COOR, SO3H
Amino Groups in EAS? How?
Cannot add NH2 directly, must first use Nitrate (NO2) and reduce (Zn/HCl or Fe/HCl to get aniline)
If you have an ortho/para director and a meta director on the same ring, where do you put the group?
strongest activator determines where the third group goes
How can substituents change the ring’s reactivity?
2 Competing Forces
Inductive Effect: moving electrons through sigma bonds based on electronegativity (Cl pulls electrons away via induction, or COR does as well since oxygen pulls on C, creating carbocation needing electron density that pulls on ring)
Resonance Effect: moving electrons through pi bonds via lone pair donation (OCH3 pushes electrons into ring via resonance)
Which competing effect is stronger for ring reactivity?
In most cases, except halogens, resonance is stronger than induction.
What do classification of substituents reveal?
How fast the ring reacts and where the electrophile goes (ortho/para or meta)
Strong Activators
NH2, NHR, OH, OR: lone pair directly next to ring, so they can do resonance and donate electrons to ring
Moderate Activators
Amides (NHCOR) and Esters (OCOR): molecules that have a lone pair but also a carbonyl; lone pair is busy resonating with carbonyl so they give slightly less to ring, so they are slightly less activating
Weak Activators
Alkyl groups (—R), donating electron density (partially) via hyperconjugation and inductoin
Strong Deactivators
NO2, CN, SO3H, CF3
Moderate Deactivators
Carbonyls (aldehydes, ketones, esters, acids)
Exception
Halogens (—F, —Cl, —Br, —I), deactivators slowing the reaction via induction but they are ortho/para directors because they are electronegative but have lone pairs that stabilize the sigma complex through resonance (no positive charge pulling stuff out causing two positive charges to NOT be next to each other)
Priority Rules for a Group if There Are Competing Effects on Existing Ring
Reinforcing Effects: if both groups direct to the same spot, major product
Opposing Effects: strongest activator wins the argument
Ranking: NH2>OH>OR>R>Halogen>Meta Directors
Steric Rule: substitution rarely occurs at the position between two groups that are meta to each other bc it is too crowded
What happens when you use groups like NH2 and OH on the ring?
Super electron rich, make ring super reactive, so if you try to brominate it, the reaction is so fast it won’t stop at one bromine, but 246 tribromoaniline.
How do you address the issue of incredible ring activity to prevent trisubstitutions
React the nitrogen (NH2) group by reacting it with acetyl chloride to form an amide
How does making an amide tame the substituents?
The lone pair on the nitrogen is now distracted because it is also resonating with the new carbonyl group (moderate substituent), so that you can add one bromine usually para
How do you preserve the original NH2?
Acid and water (H3O+) to chop off the acetyl group to get original NH2 back
How do you synthesize a para product vs. meta product in the case of NH2?
Para: add the ortho/para director first, adding the bromine first, when you add the nitro group, bromine forces it into para position
Meta: add meta director first, the NO2 group, then, when you add bromine, the nitro group forces it into the meta position
What does Friedal--Crafts Alkylation Do?
Adds an alkyl group (R)
What are the limitations of Friedal-Crafts alkylation reactions?
Carbocation rearrangements: electrophile is a free carbocation. If a primary or secondary carbocation can rearrange to a more stable secondary or tertiary one via a hydride or methyl shift, it will do so before attacking the ring
Example: You cannot make n-propylbenzene using 1-chloropropane, you will get isopropylbenzene instead
Polyalkylation: alkyl groups are activators. Once a group is added, the ring becomes more reactive than the starting material (inductive effect of donating electron density).
Example: Reaction doesn’t stop at one addition, as it gets more activated as you go along. To prevent this in synthesis, you must use an excess of benzene (so electrophile reacts with bromine than likely for benzene)
Friedal Crafts (Both Alkylation and Acylation) fail if the ring is already substituted with a moderate or strong deactivator (like NO2, or SO3H)
Takeaway: if you need to add an alkyl and nitro group, you must add the alkyl group first.
What do we do instead to combat this “problematic” alkylation reaction issue?
Nucleophilic Substittuion
What do you do to block the para position and make sure that the groups add ortho?
SO3H is bulky and para directing (ortho would lead to steric hindrance), so add SO3 and H2SO4 to install an SO3H group and block the para position, and add your desired group to the ortho position. Use H+, steam, and heat to remove the blocking group after.
This is for when you want an ortho-disubstituted product but the first group normally directs to the para position
Nucleophilic Aromatic Substitution (NAS)
Opposite of Electrophilic Aromatic Substitution
In NAS, Ring=Electrophile, and nucleophiles are (-OH, or -NH2) attacking it
2 Main Kinds:
Addition-Elimination Mechanism (SnX)
Elimination-Addition Mechanism (Benzyme)
Addition-Elimination Mechanism (SnX) Overview
One of 2 ways to perform electrophilic aromatic substitution
Addition-Elimination Mechanism: Negative Ring
Reactant: A good leaving group on a benzene (halogen) and strong withdrawing groups like NO2, CN, and CF3 positioned ortho/para to the leaving group to soak up the negative charge (NOT meta!!!)
Intermediate: Meisenheimer Complex (resonance-stabilized anion)
Reagent: NaOH or NaNH2
Product: Replacing leaving group with the strong base component like NH2 or OH
Addition-Elimination Mechanism (SnX) Mechanism
Addition (slow step) nucleophile attacks the carbon attached to the leaving group, creating a negatively charged intermediate. NO2 is needed because this negative charge is passed around the ring and lands directly on oxygen atoms of the nitro group via resonance to stabilize the intermediate
Elimination (fast step): double bond reforms, and the leaving group (halogen) is kicked off
Meisenheimer Complex
Resonance stabilized anion, opposite of Sigma complex, which is a resonance stabilized cation
Elimination-Addition Overview (Benzyne)
Triple Bond
Reactant: Halobenzene
Reagent: very strong base like NaNH2
Product: mixture of 2 products, as nucleophile can add on either side of the triple bond (cine and ipso products), and a relatively equal amount of para and meta
Regiochemistry: because the nucleophile can attack on either side of the triple bond, we get a mixture of isomers (ortho/meta products)
Ispo and Cine
Ipso Substitution: whe the nucleophile ends up on the same carbon that originally held the leaving group
Cine Substitution: nucleophile ends up on the carbon adjacent to where the halogen was
Elimination-Addition Mechanism (Benzyne)
Step 1: Elimination: the strong base pulls a proton H+ off the carbon adjacent (ortho) to the halogen, creating a triple bond inside the benzene ring, aka the benzyne intermediate (benzyne is angry because triple bonds want to be 180 but they are 120 int he ring)
Step 2: Addition: nucleophile immediately attacks one of the two carbons of the triple bond, breaking it back into a double bond
Since the nucleophile can attack on either end of the triple bond, you often get a mixture of products (cine) substitution, where the nucleophile ends up on the carbon next to where the halogen started
When do you use Friedal Crafts Alkylatoin vs. Acylation?
If you want a straight chain, use acylation followed by Clemmensen or Wolff-Kisher reduction to avoid the rearrangements of alkylation
When do you use NAS? As opposed to EAS?
If you see a strong nucleophile like NaOH or NaNH2 on the arrow, and a halogen/good leaving group on the ring
When do you use benzyne NAS vs. SnX NAS?
If there are no EWGs, think Benzyne. If there are nitro groups ortho/para, think SnX.
Gatterman-Koch Synthesis
Adds an aldehyde directly to the ring
Reactants: Benzene
Reagents: CO, HCl, and AlCl3, CuCl
Products: Aldehyde attached benzene
Clemmensen Reduction
Converts a ketone or aldehyde group (C=O) into a simple alkane group (—CH2—), eraser in orgo
Commonly used after friedal crafts acylation to turn the acyl group into an alkyl group, bypassing the rearrangement problems common in Friedal Crafts alkylation.
Reactants: Aldehydes or ketones (specifically targets the cabronyl carbon of aryl or aliphatic (ring or aromatic ring) ketones/aldehydes
Reagent: Zn(Hg) and conc HCl
Product: Acyl turns into alkyl group