ceutics distribution of drugs (+ oral PK review)
1/61
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
62 Terms
phase 1 vs phase 2 metabolism
phase 1: oxidation rxns-> make a more hydrophilic substance
phase 2: conjugation-> attach a polar molecule to the compound
oral bioavailability equation

what is the major site for absorption?
a. stomach
b. duodenum
c. jejunum/ileum
d. colon
c. jejunum/ileum
(colon is for water reabsorption)
what characteristics make the SI a great site for absorption
luminal folds, villi, microvilli
what is dissolution and why is it important
- process by which a drug moves from solid state into solute
(ex: capsules-> dissolves into solution)
- drug MUST be in solution to be absorbed
t/f: only ionized drugs typically cross the membrane
false. nonionized
t/f: first pass extraction can occur in both the gut wall and the liver
true
what source of energy does pgp use
ATP hydrolysis
what can make a substrate for pgp more susceptible to efflux
- any parameter that slows down permeation through membrane
1. more lipophilic drug (a hydrophilic drug probs wont pass through membrane and wont contact pgp)
2. higher molecular weight drug (slower permeation)
caco-2 cells
- commonly used in the pharmaceutical industry to test human intestinal permeability and bioavailability of orally administered drugs
- human colon carcinoma cells that exhibit normal intestinal tissue traits
what are the 3 compartments of body water and what is the majority
1. intravascular= in the blood (7.1%)
2. interstitial= in the tissue (38%)
3. intracellular= cytoplasm of tissue cells (55%= majority)
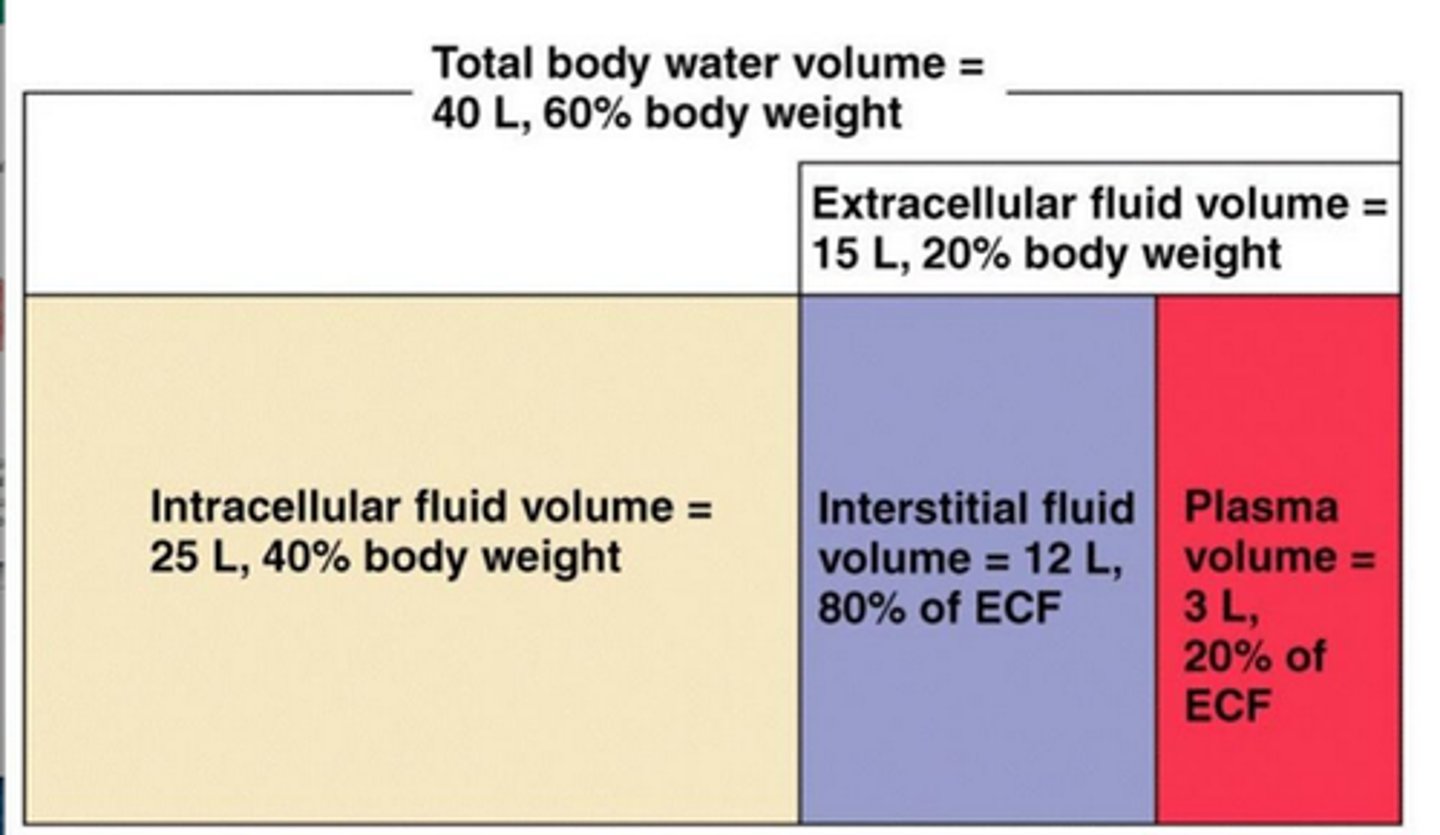
for an average 70kg person, about how many liters of water are in the body?
how much of it is plasma?
ECF?
ICF?
about 42 liters of total body water
(3L plasma, 16L ECF, 23L ICF)
what does it mean when we say a drug has a high volume of distribution
instead of just being confined to intravascular space, it reaches interstitial tissue and intracellular
ex: ethanol-> small organic molecule distributes to all sites
the total volume of fluid compartments to which drugs may be distributed is about _________ L in an adult
42L
is a drug that has a high volume of distribution more likely to be lipophilic or hydrophilic
lipophilic because can distribute through tissue and cells
what is the apparent volume of distribution and what can it tell about a drug
- measures drug distribution; drugs will distribute to parts with no body water such as hair/bones/nails too
-highly lipophilic drug will therefore have VERY high volume of distribution and SLOW elimination rate since it is sequestered in bone/fat
- this is not a true physiological space
a highly lipophilic drug would have a _________ volume of distribution and _________ elimination rate
highly lipophilic drug will therefore have VERY high volume of distribution and SLOW elimination rate since it is sequestered in bone/fat
describe what the following indicates regarding a drug
small volume of distribution (ex: 5)=
med volume (ex: 10-40)=
large (ex: 40-50k)=
(note avg body water is 40-42)
small= mainly stays in plasma
med= similar in plasma and tissues
large= mainly in tissues, not really in plasma
if drug X has a volume of distribution of 1L/kg, where would we expect this drug to be in a 60kg adult?
1L/kg means 1L*60kg= 60L
since 60L> avg amount of water of 40L, we would expect this drug to be intracellular, and not solely confined to intravasc and tissues
t/f: all tissue compartments are equal and contain an equilibrium with blood
false.
there may be compartments with tissues that the drug spreads "instantly in" (denoted as compartment 1)
and there are compartments that have slower distribution (denoted compartment 2)
drug A is an oral preparation that is mostly eliminated in feces. which tissue compartment would this compartment distribute to
if its eliminated in feces that means it doesnt reach blood/ never absorbed. therefore it doesnt distribute to ANY tissue compartment
how does plasma protein binding affect volume of distribution
increased protein binding means a higher amount of drug in plasma, and a LOWER volume of distribution
how does tissue affinity affect volume of distribution
increased tissue affinity means lower concentration of drug in plasma, and a HIGHER volume of distribution
what would the distribution of a non-absorbed drug be
nothing. distribution is the delivery of a drug from blood to tissue. if its not absorbed then its not in the blood to begin with. will be in feces
t/f: absorption is necessary for drug distribution
true
how does capillary permeability affect the rate at which a drug penetrates tissues and other bodily fluids
lower capillary permeability lowers rate of distribution
(usually good except for BBB)
how does blood flow affect the rate at which a drug penetrates tissues and other bodily fluids
increased blood flow increases rate of distribution
what explains why some basic drugs can be shifted back to the stomach post absoprtion
due to pH differences. pulled back into stomach and can become ion trapped
(can also happen in urine)
if a basic drug is in the bloodstream, but then shifts and becomes ion trapped in the stomach due to pH differences, how will this affect the ct curve
shifts down bc drug WAS absorbed but stomach pulled it back in so less drug is in blood
which factors can affect transporters at the blood/tissue junction and therefore affect distribution
1. saturation
2. competition
-> both can slow the rate of distribution
how does a more rapid perfusion rate affect distribution towards equilibrium
greater perfusion rate= tissue has a lot of blood going through it (perfusion)= rate of equilibrium is faster= shorter time period
which parameters does the equilibrium between drug in plasma and drug in tissue depend on
plasma: Vp* C
[volume of plasma* drug concentration]
tissue:
VtKpC
[volume of tissue affinity constant for drug to tissue concentration of drug]
summarize how the following factors can affect the rate of distribution
- perfusion
- tissue membrane permeability
- plasma proteins
-pH differences
1. perfusion: greater perfusion= faster rate
2. tissue membrane permeability: lipophilic drugs= faster
3. plasma proteins: more bound means stays in plasma= slower
4. pH differences: weak acids trapped in bases, and weak bases ion trapped in acids
which organs are the most perfused in the body? how does this correlate to time of distribution?
liver, kidney, brain
+ skeletal muscle when exercising
-> more perfusion= shorter time of distribution bc greater rate
which drugs have no barrier to distribution via tissue membranes and are SOLELY perfusion limited
lipophilic drugs
which will have the shortest time to equilibrium in tissue for a lipophilic drug
a. brain
b. fat
c. bone
brain bc highly perfused. fat will sequester the drug but bc its so poorly perfused it will take a while to reach equilibirum
t/f: well perfused tissues take up more drug than poorly perfused tissues
true. more perfused tissues will be exposed to more drug molecules per unit of time
what is Kp? how does it relate to the amount of drug in tissue?
Kp is the equilibrium constant (or affinity constant/ permeability constant)
Kp= Ct/Cv or concentration of drug in tissue divided by concentration of drug in vein
makes sense= if this is the affinity constant than ur comparing how much of the drug is in the tissue to how much was not retained/coming out
what can the amount of drug in tissue be expressed as
Vt* Ct (aka tissue volume times tissue concentration)
OR
VtKpCv (aka tissue volume times equilibrium constant times venous concentration)
remember Kp is just Ct/Cv so it cancels out
explain the concentration of drug in artery, tissue, and vein, using the equations
artery: takes drug to tissue= Ca
tissue: VtKpCv [aka tissue volume perfusion constant venous concentration]
vein: Cv-> volume of drugs not taken up by tissue
(why? maybe less affinity, maybe equilibrium is already established)
rate of blood flow in artery and vein (Q) is the same!! thats why we ignore
tissue: Vt and Ct
what is the fractional rate of exit of a drug from tissue expressed as
kT= rate of exit/ amount in tissue
tissue distribution half life is expressed as
half life= 0.693/kT
orrr
[(0.693 Kp)/ (Q/Vt)]
remember kT is the fraction rate of exit= (rate of exit/ amount in tissue)
time it takes for drug concentration to decrease by half- either in blood or tissue
![<p>half life= 0.693/kT</p><p>orrr</p><p>[(0.693 Kp)/ (Q/Vt)]</p><p>remember kT is the fraction rate of exit= (rate of exit/ amount in tissue)</p><p>time it takes for drug concentration to decrease by half- either in blood or tissue</p>](https://knowt-user-attachments.s3.amazonaws.com/3d4a3a25-d1eb-466c-82b8-ad2d07c51c77.png)
given tissue distribution half life= 0.693Kp/ (Q/Vt)
how would a higher affinity drug affect half life?
a lower affinity drug?
high affinity would be a larger Kp-> numerator is larger-> larger half life= drug will leave the tissue slower
low affinity would be smaller Kp-> numerator is smaller-> smaller half life= drug will leave the tissue faster
drug would leave slowly from tissues with a _____ Kp and a _____Q
high Kp (bc high affinity)
low Q (bc poorly perfused)
if arterial concentration remains constant, what would happen to tissue concentration and the rate of uptake over time
tissue concentration would increase
the RATE of uptake will decrease over time= bc less Ca since it accumulates in tissue over time
Given kidneys, brain, and fat, if there is
- a constant Ca (arterial concentration)
- same Kp (tissue affinity)
- perfusion rate= 4, 0.5, and 0.03 ( w respect)
in which tissue will it accumulate the fastest?
will accumulate in the kidney the fastest (bc q=4)
then brain, then fat
which kind of drugs are more limited by permeability rather than perfusion?
-polar and high molecular drugs are more permeability limited
- lipophilic drugs are perfusion limited bc will cross no matter what
(ex: polar molecule will reach equilibrium faster in muscles than brain bc muscle are more hydrophilic)
do lipophilic drugs reach equilibrium faster in brain or muscle? why?
brain
lipophilic drugs are perfusion dependant, brain is more highly perfused
do polar molecules reach equilibrium faster in brain or muscles? why?
muscles bc permeability limited
muscles are more hydrophilic and the brain is lipophilic (BBB)
thiopental takes 1.4min to reach 50% of equilibrium value in CSF while salicylic acid takes about 115min.
thiopental:
partition coeff= 3.3 (lipophilic)
pKa= 7.6 (basic)
salicylic acid:
partition coeff= 0.12 (hydrophilic)
pKa=3.0 (acidic)
what can explain the difference in tissue distribution?
1. partition coefficient
-thiopental is very lipophilic. only depends on perfusion rate.
-salicylic acid is hydrophilic so it is also permeability limited. brain is more lipophilic
2. acidity
normal ph is 7.4. thiopental is unionized which favors membrane permeability. salicylic acid will be ionized, making it less permeable
describe a perfusion limited schematic
compartments?
depends on?
what kind of drugs?
1 compartment= drug moves easily from blood to interstitial fluid to cell membrane to intracellular fluid. high Kp
- dependant on blood flow; more drug must be administered (no limitations/resistance)
- for LIPOPHILIC drugs
how does a permeability limited schematic at the cell membrane correlate to equilibrium, perfusion, and changing drug concentration
-if permeability limited then slower movement into target cell-> takes longer to reach equilibrium
- independent of perfusion and changing drug concentration; will only take in so much per unit of time
t/f: a perfusion limited drug will have a higher therapeutic effect if drug concentration is increased, but a permeability limited drug will not alter its effect
true. permeability limited will only take in so much per unit of time. changing concentration will not force in any more drug molecules
describe a permeability limited schematic AT THE CELL MEMBRANE (not capillary)
potential reason?
- blood is taking drug to tissue but there is resistance into target cell bc
1. not permeable membrane or
2. chemical property of drug
describe a permeability limited schematic at BOTH the capillary and cell membrane
where will this happen?
- slow movement from blood to tissue AND slow from fluid to membrane
1. CNS (BBB)= capillaries are not fenestrated and podocytes prevent drug from leaving capillaries
2. placenta
a ________ Kp suggests high tissue affinity, while a _________ Kp suggests greater plasma protein affinity
a. higher, lower
b. lower, higher
c. not applicable
a. higher, lower
higher Kp= tissue affinity
lower Kp= plasma protein affinity
eyes, fat, and bone have a ________ rate to equilibrium due to ________ perfusion
longer rate to equilibrium; low perfusion
which 4 factors lower BBB permeability
1. lack of pores btwn capillaries
2. endothelial cells contain ATP-dependant transport (not everything is absorbed)
3. capillaries are surrounded by astrocytes
4. low protein concentration in interstitial fluid (no plasma proteins= no affinity)
but purely lipophilic drugs can get past!!
what can increase the permeability of BBB? which drugs may get past it?
1. inflammation
2. underdeveloped at birth
-> highly lipophilic drugs can get past
if a drug is very hydrophilic, it will not get past the BBB and instead result in __________ Cv and _____ Kp than when compared to lipophilic drug
higher Cv, low Kp
bc not getting absorbed by tissue (brain), but instead kicked out into vein
t/f: even if drugs successfully cross into the brain, it is often at a slow rate and the drug concentration never reaches adequate therapeutic levels
true
t/f: the placenta is an absolute barrier for drugs due to its multiple layers of cells btwn mother and fetus
false. latter part is true but it is NOT an absolute barrier