Heme #2: Exam #3
1/74
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
75 Terms
Chronic Lymphocytic Leukemia
Chronic Lymphoproliferative Disorders
-clonal proliferation disorders of MATURE B or T cells
-insidious onset and an indolent course
-subclassification depends on morphology, immunophenotyping, cytogenetics, clinical features, and molecular testing
lymphoma
disorders that affect lymph nodes and other extramedullary sites
Chronic Lymphocytic Leukemia (CLL)
-most common chronic lymphoproliferative disorder
-generally a neoplasm of mature B lymphs but can involve T cells
-B-Cell CLL is what we focus on
->50 years old
-lymphocytosis >5
-small, mature lymphocytes with soccerball chromatin
-SMUDGE CELLS (basket cells) on PBS
-lymphoid accumulation crowds out normal bone marrow elements
-infiltration of malignant cells in the lymph nodes and spleen (so you get lymphadenopathy or splenomegaly)
-reduced humoral immunity
Small Lymphocytic Leukemia (SLL)
-synonymous wiht CLL but tends to involve lymph nodes and other lymphoid organs
-it will have the same tissue morph and immunophenotype but without the PBS and BM leukemia
Etiology of CLL
-no specific agent
-accumulation of dysfunctional mature lymphs in PBS and BM
-failure in apoptosis causing survival and expansion of the malignant clone
-normal BM elements are replaced causing anemia, thrombocytopenia, and neutropenia
Clinical Features of CLL
->50 in males more
-gradual onset
-routine physical CBC is where it is found
-lymphocytosis, lymphadenopathy, splenomegaly
Lab findings in CLL
-lymphocytosis >5
*mature lymphs with cracked chromatin/soccer ball look
*round to slighly indented nucleus
*scant cytoplasm
*indistinct or absent nucleoli
**prolymphs are sometimes seen
-SMUDGE CELLS are diagnostic for CLL
-N/N anemia sometimes
-normal to low reticulocytes
-thrombocytopenia sometimes
-WBC INCREASED
BM in CLL
-mature lymphs
BM INFILTRATION:
*Nodular
-distinct, randomly distrubuted lymphoid aggregates
*Insterstitial
-lymphs infiltrate interstitium without displacing fat cells
*Diffuse
-entire BM is replaced with lymphs
*Combination of some of these
Other lab results
-hypogammaglobulinemia in most patients
-IgA, G, M (GAM) decreased bc B cell disease and abnormal function
-leads to bacterial and viral infections
-can lead to autoimmunity like AIHA (autoimune hemol), ITP, pure red cell aplasia, etc.
-10-30% will develop AIHA so you may see spherocytes and polychromasia, nRBCs, positive DAT, increased bilirubin
**look for SPHEROCYTES and REPORT PRESENT OR NOT LIKE WE DO WITH SCHICHTOCYTES!!
Smudge Cells
-CLL
-fragile lymphs that break during slide prep
-can be removed by mixing 1 drop of albumin with one drop blood before PBS made
-this needs to be done for an accurate lymph count
-but albumin distorts red cells so need original smear too
-plt estimate on original slide too (could falsely lower plt count bc diluted)
Immunologic features of CLL
-can't distinguish B vs T cells on PBS
-monocloncal antibodies and flow helps
-these cells don't mature to the final stages of B cell maturity
-weak IgM/D with kappa or lambda light chains
-CD19+, 20, 22, 79a, 5 (USUALLY SEEN ON T CELLS), CD23, 43
-you may see reactive-like lymphs to flow is important on anyone over 50 with lymphocytosis >5
CLL genetic abnormalities
-50-60% have
-FISH!
-Trisomy 12 (poor prognosis)
-del or t(13q14) (longer survival)
-11q22-23 del (poor)
-17p13 del (biggest predictor of POOR!)
CLL Survival
-4-5 years
-many pts asymptomatic (indolent)
-treatment not started until pt has a lot of lymphocytosis, splenomegaly/lymphadenopathy, anemia, neutropenia, thrombocytopenia, autoimmune events, infections
-sometimes CAN be aggressive 1-2 year survival
-prognosis based on lymphcyte doubling time, bone marrow infiltration patterns (diffuse is worse), and chromosome abnormalities
-not curable with current tx but long survival ish
CLL transformation
-can progress when a new pop of malignant lymphs develops
-increases resistance to therapy
-prolymphocytic leukemial, diffuse large B-cell lymphoma (Richter syndrome), or acute leukemia (not often)
Richter's Syndrome
-CLL converts
-progressive lymphoma
-fever, weight loss, organomegaly, worsening cytopenias
-very poor response to chemo
(CLL): ZAP-70
-this and CD38 are usually positive when there is unmuated Ig variable regions
-more high risk
-NEGATIVE PROGNOSIS!!! for CLL
PLL (prolymphocytic leukemia)
-proliferation of malignant PROlymphs (b or t)
-CLL can transform or it can occur separately
-extremely rare but more in 60+ year olds with males
B-PLL
-WBC >100
-Absolute lymphocytosis
-marked splenomegaly NO lymphadenopathy
-anemia and low plts
->55% cells are prolymphs
Prolymphs
-9-18 microns (large cells)
-round or indented nuclei
-mature, condensed between blast and lymphcyte chromatin
-0-1 prominant nucleoli
-large amounts of basophilic cytoplasm
-NO GRANULES!
-punched out looking nuceloli surrounded by a halo of condensed chromatin
B-Cell PLL
-sIg+, 19,20,22,79a,79b,5
-NO CD23
T-Cell PLL
-2,3,5,7,tdt-, 4+,8- mostly (could be both pos or both neg)
PLL genetics
-chromosome t or inv 14 q11 q32-
-t 11;14 q13q32 (worse prog bc response poor to tx, short survival)
CLL vs OTHERS
-acute lymphoblastic leukemia (ALL)
-reactive lymphocytosis
-PLL
-non-hodgkin lymphoma (mantle cell lymphoma)
-t-cell large granular lymphocytic leukemia
-sezary cell lymphoma
-hairy cell leukemia
Hairy Cell Leukemia!!!
Hair Cell Leukemia summary
-uncommon chronic B-cell lymphoproliferative disorder
-irregular HAIRLY cytoplasmic projections on the malignant cells
-2% of leukemias
-middle aged males 4-7x more than females
-52 is median age very rare in less than 30 year olds
causes of HCL
-ionizing radiation
-organic chemicals
-farming
-wood working
-EBV
origin of HCL
-clonal expansion of mature B cells with light chain resitricted surface Ig expression
-antigen-experienced memory B cell from the post-germinal center
-usually have some somatic mutation in their Ig variable genes
-single disease entity that is very distinct from other B-cell malignancies
Clinical Findings of HCL
-1/4 asymptomatic
-abdominal pain/fullness
-massive splenomegaly
-hepatomegaly
-lymphadenopathy
-fatigue, weakness, weight loss, night sweats, fever
-bleeding/bruising bc low platelets
-recurrent infection bc neutropenia
Lab Findings in HCL
-pancytopenia bc of splenomegaly and BM failure
-SPLEEN CAUSES PANCYTOPENIA BC:
*sequesters 90% of platelets
*30% of RBCs, and 65% of granulocytes
-LEUKOPENIA! with neutropenia and monocytopenia
-WBC are <4
-infections are the leading COD with HCL
-N/N anemia and plts 20-100
-azotemia (uremia) is a poisonous condition caused by failure of the kidneys to remove urea from the blood
-hypergammaglobinemia (TOO MANY ANITBODIES)
-abnormal liver function tests!!
PB and BM of HCL
PB:
-nucleus is large, oval, reniform (kidney shaped round or slightly clefted), eccentric but may be central
-chromatin homogenous and relatively fine
-cytoplasm is abundant pale with FINE PROJECTIONS (hairy)
-can be vacuolated
-HC are not definitive diagnosis!! bc it can look like splenic marginal zone lymphoma!!
BM:
-hypercellular mostly
-fibrotic so dry tap bc hairy cells infiltrate the marrow
-honeycomb apperance due to the cytoplasm making a halo around the nucleus
-hairy cells can look like a FRIED egg
-reticulin stain is always increased reticulin fibers
Cytochemical Features of HCL
-TRAP positive
-acid phosphatase isoenzyme #5 still works with tartrate added
-with and without tartrate serves as + and - control
-you need 40+ granules in at least 2 cells to be called + *maybe just 1 cell
-staining will be red/purple granules in the cytoplasm
-Neutrophils negative
Immunophenotyping in HCL
-bright CD19
-20,22
-also for uncommon ones like CD11c usually on monocytes and neutrophils
-CD25 usually on activated T-cells
-Extensive co-expresion of CD11c + CD22 and CD103 are highly sensitive for HCs
-
HCL Variant
-more aggressive form with median age of 71
-cell is smaller, round nucleus, prominent nucleoli, higher N:C ratio, basophilic cytoplasm with rare projections
-WBC may be >50
-cytopenias and splenomegaly
-CD25 and maybe CD103 negative
-poor prognosis
treament of HCL
-usually not tx until significantly symptomatic
-watch and wait usually
-tx started with significant neutropenia, anemia, low plts, symptomatic splenomegaly, recurrent infections, night sweats, fever
TX NOW:
-splenectomy for normal counts
-interferon increases prognosis by inducing apoptosis in nonadherent hairy cells
-nuceloside or purine analogs target lymphs and are cytotoxic to resting and dividing cells (pentostatin, cladribine)
-many relapse but can use the same tx again
-with each tx though the disease free time is shortened
Experimental TX:
-monoclonal antibodies against CD20 (rituxan)
-BL22 or anti-CD22 anitbody
-anti-CD25 antibody
prognosis of HCL
-median survival rate of 4-7 years before interferon and nucleoside analogs. now 10+ years
-infection is the greatest risk of death
Starting Plasma Cell Neoplasms!!!!!!!!
plasma cell neoplasms general info
-M-spike on protein or urine electrophoresis (bc of monoclonal antibodies being made
-multiple myeloma, plasmacytoma, MGUS, waldenstrom's macroglobulinemia, primary amyloidosis
plasma cells are
-fully mature B cells that have undergone activation by antigens
-4% of BM and NOT usually in PBS
-in response to infections, inflammatory or malignant conditions
-coarse chromatin 8-20 microns in size; ECCENTRIC nucleus with 80s prom dress cytoplasm. no granules. deeply basophilic cytoplasm
-have haf a HOF or clearing of golgi apparatus
types of antibodies made by plasma cells
-GAMED
-each has 2 light chains and 3 heavy chains
HEAVY CHAINS:
-alpha, gamma, Mu (micro), epsilon, delta
LIGHT CHAINS:
-kappa, lambda
plasmacytoid lymphocyte
-between lymph and plasma cell
-15-20 micorns, chromatin less clumpled, visible single nucleoli, eccentric or central nucleus but deeply basophilic cytoplasm
Multiple Myeloma
-most common plasma cell neoplasm
-numerous lytic bone lesions
-median age 66 with male predominance
-~20% asymptomatic
-bone pain or fracture due to lesions
-fatigue, thirst, nausea, infection, weakness, numbness, renal insufficiency
-bone radiographs diagnose osteolytic lesions, osteoporosis, etc.
cause of MM
not well known but:
-atomic bomb survivors, organic solvents, toxins in textile industry, chemicals, pesticides, herbicides
pathophysiology of MM
-acceleration of plasma cell growth due to genetic alterations that prevent normal differentiation and apoptosis
(IL-6 is secreted with accelerates plasma cell growth and inhibits apoptosis)
-activation of bone resorption factors or osteoclasts
(OAF (osteoclast activating factor) is secreted and stimulates osteoclasts to absorb bone); serum calcium increases due to bone loss)
-production of abnormal monoclonal protein
(as production of plasma cells increases, the production of immunoglobulin increase)
osteoclasts
-large cells up to 100microns
-low N:C ratio
-several, separate round nuclei
-blue, slighly granular cytoplasm
plasma cell tumors may go to
other parts of the body besides BM
PBS in MM
-N/N anemia
-ROULEAUX (STACKING) OF RBCs
-abnormal proteins in the blood cause them to stick
-plasma cells may be seen
-cytopenias may form bc plasma cells are replacing normal BM cells
-increased protein caused by excess Igs causes blood smears to have a blue ish background!!!
-elevated ESR meaning quicker settling because of rouleax
-as plasma cells take over, low plts and anemia, and neutropenia occur
BM in MM
->30% plasma cells usually in sheets/large aggregates
-could be abnormal looking with fine chromatin, nuceloli, or intracellular inclusions with Igs
-mott cells, flame cells could be seen
-normal lymph and plasma cells decreases so less antibodies around meaning more risk of infection
Flame Cells
-reddish purple cytoplasm
-more Ig than usual for sure
Mott Cells
-plasma cell filled with globules containing Ig called RUSSELL BODIES
-grape cells
Dutcher Bodies
-inclusion in plasma cells seen in MM sometimes that have GLYCOGEN!!!
-usually in neoplastic plasma cells
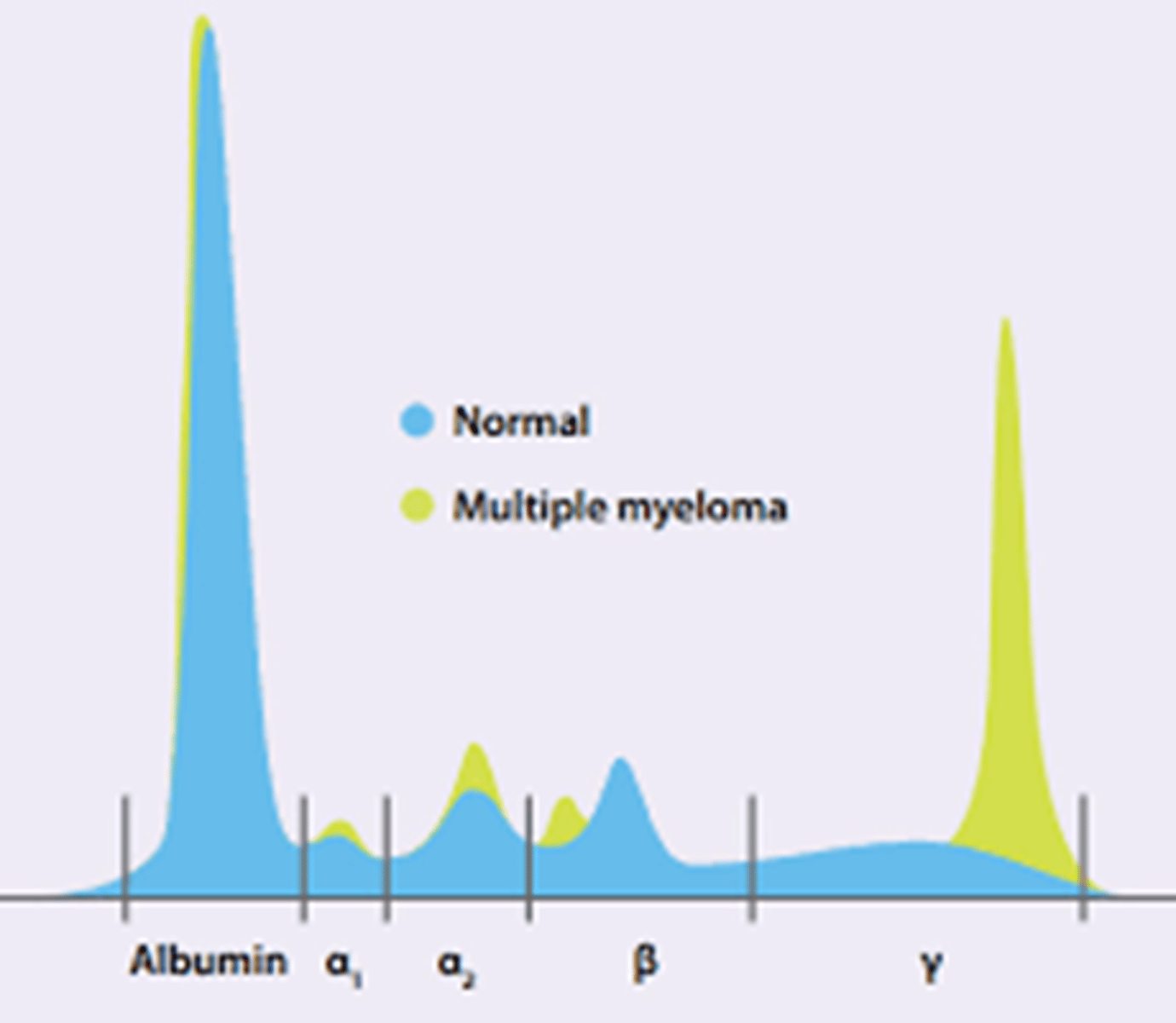
serum protein electrophoresis for MM
-monoclonal proteins can be confirmed
-Ig, alumbin, and minor proteins
-ALBUMIN, alpha 1, 2, beta, ,gamma (where MM is)
-M SPIKE = multiple myeloma
-these monoclonal proteins have only ONE light chain (kappa or lambda) and one heavy chain (GAMED)
-can see the m spike in serum or URINE
Bence Jones proteins
-excess free light chains in the urine
-serum electrophoresis is normal
-Bence-Jones Proteinuria
-small proteins that can be filtered by the kidney but cause kidney damage
immunophenotyping in MM
-lack sIg
-no 19,20
-have 138, 38, 56+ (usually)
cytogenetics in MM
-hard because plasma cells don't readily divide to produce metaphase cells
-FISH!
-many chromsome abnormalities are found (not gonna learn)
prognosis and tx of MM
-poor prognosis
-determine prognosis with labeling index test
-plasma cell labeling index (PCLI) is a fluorescent slide technique to identify plasma cells in the PBS and BM and report proliferation index as a prognostic indicator of myeloma
FAVORABLE:
-t(11;14)
-hyperdiploidy
UNFAV:
-del(17q)
-monosomy 13
other factors:
-renal failure, Hg levels, calcium levels, B2 microglobulin levels, lytic bone lesions, % of plasma cells, amount of M-component
tx of MM
-radiation for relief of bone pain
-chemo
-autologous BM transplant
-stem cell transplant
-thalidomide
-interferon alpha
-relapse occurs in almost all patients
-infection is one of the major COD
plasmacytoma
-localized plasma cell tumors causing monoclonal spikes of SPEP
-lesions could develop of bones and develop into MM
-respiratory tract plasmacytomas have a good prognosis
plasma cell leukemia
-a complication seen in MM
-as plasma cells take over BM they spill into PBS causing increased calcium and plasmacytomas with bone pain
DIAGNOSED:
->2 or 20% plasma cells
-primary is diagnosed in the leukemic phase
-secondary represents transformation of MM
-aggressive leukemia with poor prognosis
Monocloncal Gammopathy of Undetermined Significance (MGUS)
-finding an M spike doesn't always mean MM
-benign monoclonal gammopathy is this
-small M spike with small amounts of urine light chains
-<10% plasma cells in BM with normal morph
-usually asymptomatic and these are NOT THERE:
*no lytic bone lesions
*no anemia
*no hypercalcemia
*renal insufficiency NOT there
-25% can go to MM
-tx usually unecessary unless symtomatic, not usually their COD is this
Waldenstrom's macroglobulinemia
-plasma cell disorder with an overproduction of monoclonal IgM
-lymphadenopathy, hepatosplenomegaly, hyperviscosity syndrome (decreased blood flow bc of excess protein so confusion, chest tightness, blurred vision, headache, numbness of fingers)
-no lytic bone lesions or bone pain though
-anemia and pallor
-sharp narrow M-spike on SPEC
PB/BM
-mixed population of lymphs, plasma cells, and plasmcytoid lymphs
M-spike
-IgM heavy chain with kappa or lambda light chain
-large molecule IgM so increases viscosity
-IgM can also coat platelets causing inteference with clotting facotrs so thrombotic complications
Tx:
-plasmapheresis where blood is removed, plasma is drawn off and discarded, and blood is returned
-NOT CURABLE, chemo, alkylators,
-4 years survival
-cryoglobulins are abnormal proteins that precipitate or gel when cooled to 0 degreesC, and can form in these patients
amyloidosis
-when light chains (kappa or lambda) accumulate
-amyloid: fibrous protein that deposits in the organ
-that organ will have loss of function
-commonly see weight loss and fatigue
-heart: CHF
-kidneys: nephrotic syndrome or renal failure
-Gut: malabsorption
-Tongue: macroglossia
-Nerves: peripheral neuropathy
-biopsy of affected organ can diagnose
-congo RED stain
-amyloids will have apple green birefringence with polarized light
-poor survival and treat underlying cause but COD is cardiac disease, renal failure, infection, hemorrhage
PCLI
-plasma cell labeling index
-MM patients
-measures the proliferative rate of BM plasma cells using either BM or PBS
-flow cytometry can separate plasma cells and allow for specific identification and confirmation
-measured as a % of plasma cells in the SYNTHETIC phase (synthesis phase S phase of the cell cycle where they are actively making DNA)
-tests for antibodies against 5-bromo-2'-deoxyuridine which are found in actively dividing cells
-High PCLI (>=3%) means more dividing cells and a poor prognosis
-Low PCLI (<3 is a better prognosis)
plasma cell proliferation assay (PCPRO)
-uses flow cytometry to isolate plasma cells for evaluation for clonality and analyze the DNA content of the cells
-BM in ACD, Heparin or EDTA anticoagulant is used
-isolate using CD38,138, 19 and 45
-DAPI stains the DNA and allows the DNA content to be individually quantitated based on fluoresence
Cell Phases:
-G0G1: normal resting cells and have normal DNA (2n)
-Sphase: proliferating and preparing to divide, making DNA and goes to 4n
-G2M: growth right before division; cells have 4N and are ready to go into mitosis
STARTING LYMPHOMA LESSON!!
tissue equivalent to CLL and the immunophenotype for it
*this is dif from mantle cell lymphoma why
SLL (small lymphocytic lymphoma)
-same immunophenotype as CLL (5,19,20,22,23)
-dif from mantle because it has CD23 and not FMC-7
cells found in follicular lymphoma
-BUTT cells (large cells with very irregular nuclear outlines and a deep indentation/cleft of the nuclear membrane)
-most common to circulate in the peripheral blood (leukemic phase)
preceding H.pylori infection can lead to
MALT (extranodal marginal zone lymphoma of mucosa associated lymphoid tissue)
which lymphoma represents 1/3 of all pediatric lymphoma
*what does the biopsy look like
-BURKITTs
-starry sky (sky is blue nuclei of the lymphs and the stars are pale staining macrophages)
LGL Leukemia (large granular lymphocytic)
*PBS
*2 categories
PBS:
-can look like CLL but is technically T lymphs
-the lymphs with have abundant cytoplasm with AZUROPHILIC GRANULES in the cytoplasm
2 categories:
*????????????? email her!
LGL vs reactive lymphocytosis
-LGL is a leukemia so patients also have anemia, low plts, and neutropenia
-you need molecular 2,3,5,7,8,16,57 positive too in flow negative for 4 and 56
Sezary syndrome
*cell
*markers
*diagnostic criteria
-T-cell with very irregular, convuluted nuclear outlines
-CD3,4 positive but lacks CD7
-CD4:CD8 ratio >10
-patients have erythroderma (red skin) and lymphadenopathy
-sezary cells have nuclei with marked convultions (not perfectly round); kinda cerebriform (brain like)
-Mycosis fungoides (MF) is similar
-most common cutaneous T-cell lymphoma
-diffuse skin involvement and must have a + skin biopsy
-Hgb normal
-Plt Normal
-increased WBC maybe with eosinophilia
-minimal BM involvement
tissue equivalent of ALL
?????????????
Classic Hodgkin Lymphoma vs nonHodgkin
Reed-Sternberg cell (R-S) cell which has 2+ nuclear lobes containing inclusion-like nucleoli and an area of perinucleolar clearing imparting an owl's eye appearance
lymphoma cells
-deep indentations, clefts, folds
-vacuoles maybe
NewBorn smears
-increased lymphs (funny looking or 'kid' lymphs)
-normal absolute count is 2-11 on newborn
-normal absolute count up to a month is 2-17
-immature neutrophls
-polychromasia
-macrocytic reds
-MCV 98-120 is normal
-hyposplenic picture because just not fully working yet (target cells, HJ bodies, acanthocytes)
-thrombocytopenia is NOT NORMAL, schistocytes, spherocytes, and blasts are all NOT NORMAL!