Hereditary Fundus Dystrophies
1/70
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
71 Terms
Dystrophy
defective tissue which usually goes on to degenerate. Is usually congenital.
Degeneration
a retrogressive pathologic change in a previously normal tissue usually leading to impairment via degeneration. May or may not be inherited, but is not present at birth. I.e.) lattice degeneration
Electrooculogram (EOG)
electrodiagnostic test that measures the electric potential at the level of the RPE and photoreceptors. Measurement of the potential difference between the cornea and posterior pole in light versus dark setting can be used to construct the Arden ratio to determine if there is RPE disease
1.8
normal Arden ratio, the ratio between the light peak and dark peak of EOG testing
RPE, rods
A decrease in the Arden ratio established by EOG is suggestive of _____ disease and disorders affecting the ____
Electroretinogram (ERG)
electrodiagnostic test that measures the mass electrical response of the retina. Assesses the function of both cones and rods. Can be performed when the patient is dark or light adapted separating cone versus rod function.
Negative A wave
wave of the ERG indicating photoreceptor function
Positive B wave
wave of the ERG indicating bipolar and muller cell function (inner retina)
30 Hz
flicker rate of ERG which suppresses rod responses in order to isolate cone responses
Pattern ERG (pERG)
electrodiagnostic test that is used to study macular cone function and ganglion cell function
Multifocal ERG (mfERG)
electrodiagnostic test that creates a topographic map of central retina function for the cone system
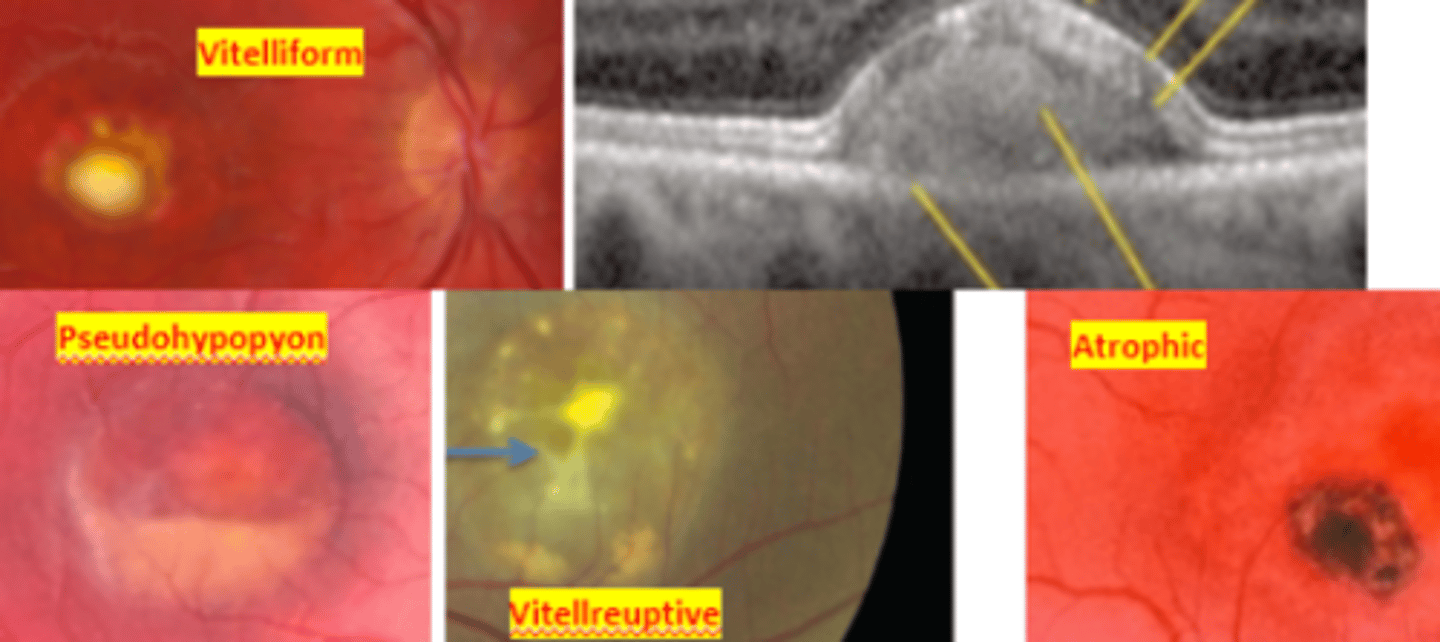
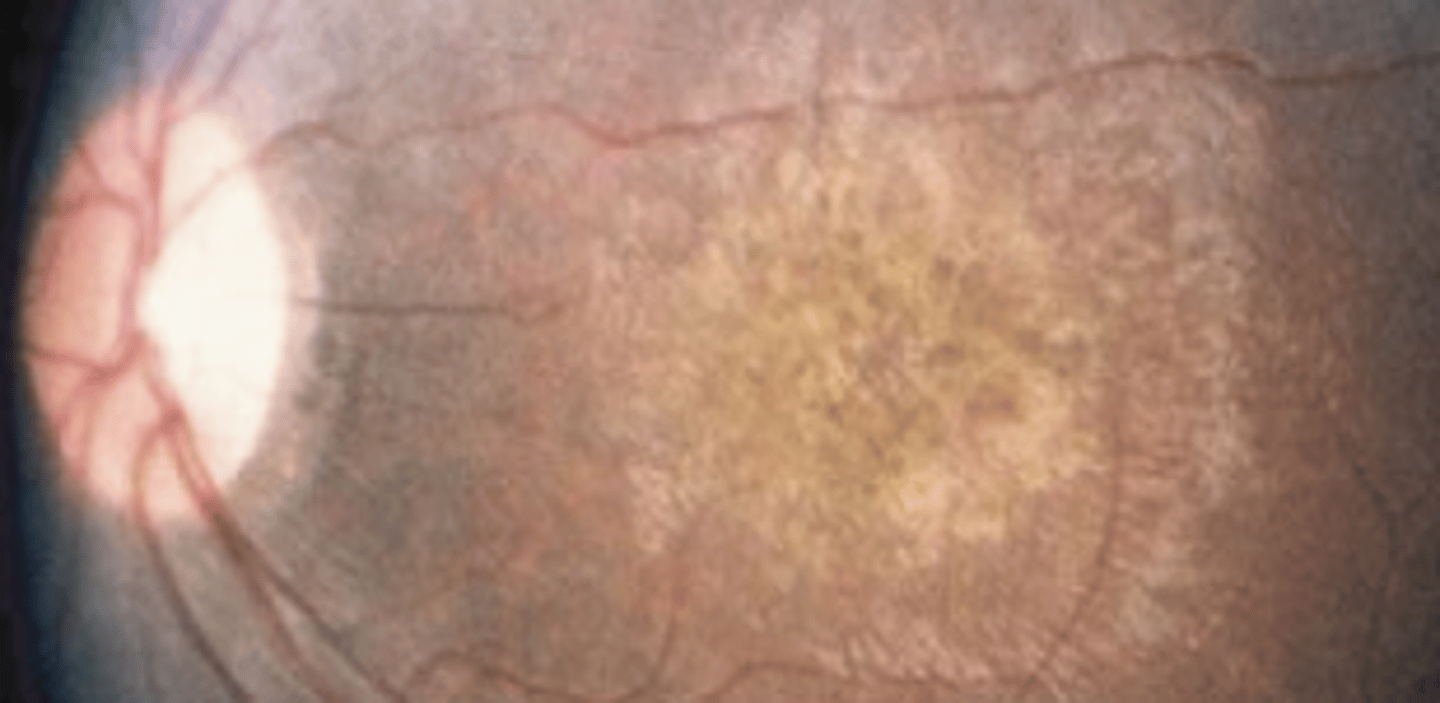
Best vitelliform macular dystrophy (BVMD)
an autosomal dominant disease resulting in mutation of the bestrophin gene. This results in bilateral subretinal ("egg yolk") deposits due to excessive lipofuscin accumulation in the RPE. Has a juvenile onset (3-15 yo). Patient will have abnormal EOG even when the fundus appears normal. ERG results will be normal. There is no treatment, however prognosis is good up until the 5th decade. Refer to low vision and for genetic testing.
1.5
Patients with best macular dystrophy will have Arden ratios below...
0
stage of best macular dystrophy where the fundus is normal, but EOG is abnormal
1 (pre-vitelliform)
stage of best macular dystrophy where there is pigment mottling at the macula
2 (vitelliform)
stage of best macular dystrophy where there are yellow egg yolk lesions at the macula (1st to 2nd decade)
3
stage of best macular dystrophy where there is a pseudo hypopyon
4 (vitelleruptive, atrophic)
stage of best macular dystrophy where there is RPE atrophy or CNVM due to break up of lipofuscin
Macular scarring, CNVM, geographic atrophy
legal blindness may occur in Best macular dystrophy is one of these three retinal events occur
Adult onset Foveomacular Vitelliform Macular Dystrophy (AOVMD)
an autosomal dominant disease presenting in the 4th-6th decade as macular lesions often centered by a pigmented spot and darkened border, possibly a milder form of best's disease. Is asymptomatic to mild often misdiagnosed as early AMD. There is no treatment, but prognosis is good unless a CNVM forms.
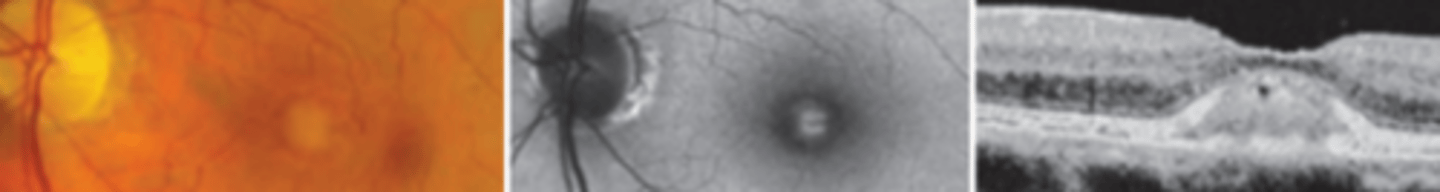
Milder presentation (EOG normal or slightly abnormal, smaller lesions), older age of onset
two factors differentiating adult onset foveomacular vitelliform macular dystrophy from Best macular dystrophy
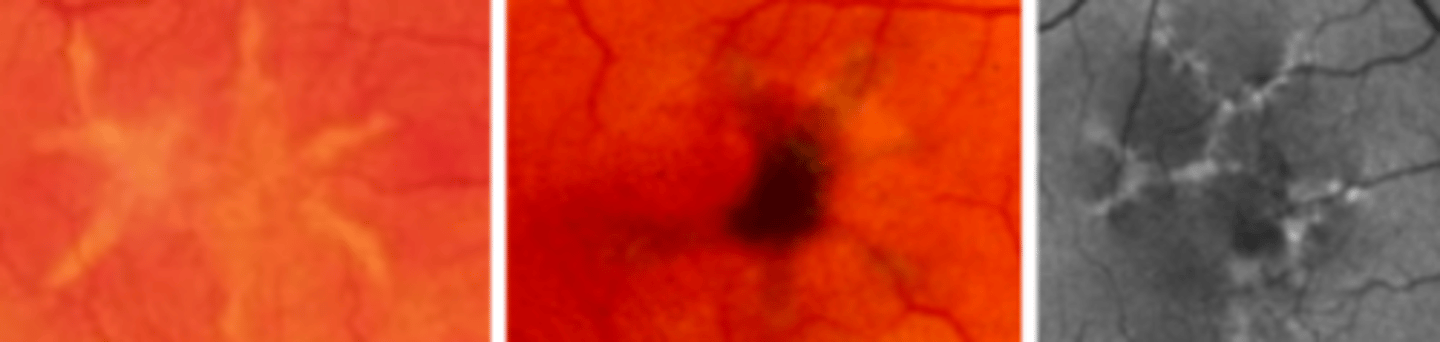
Butterfly-shaped Macular Dystrophy
an autosomal dominant disease presenting in the 2nd-6th decade as bilateral star shaped yellow lesions of the macula. Prognosis is good, CNVM and atrophic maculopathy rarely occurs. ERG may be slightly subnormal. EOG often depressed.
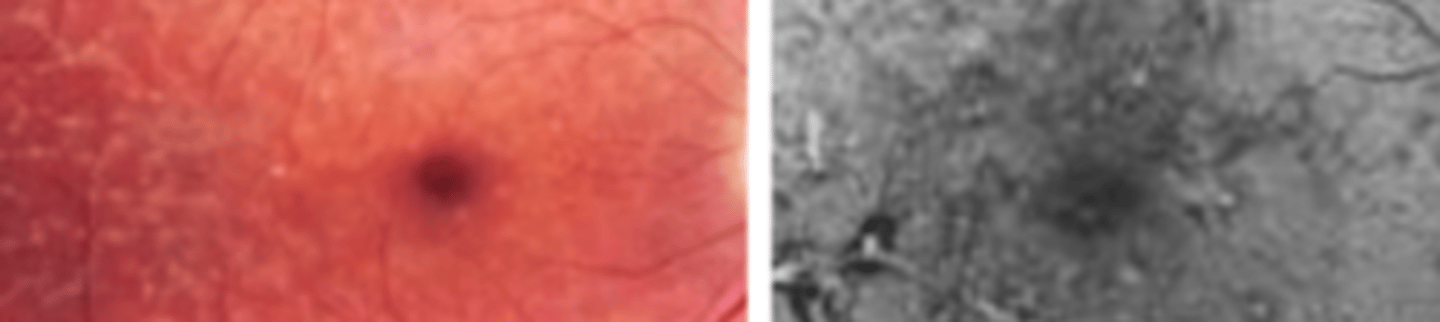
Reticular Dystrophy of the RPE
an autosomal dominant* or autosomal recessive disease presenting in the 3rd-5th decade as coarse, net like lesions of the posterior pole involving the RPE. No treatment, but may fade with age and has good prognosis. ERG normal, EOG subnormal due to RPE involvement.
Retinitis Pigmentosa (pigmentary retinal dystrophy)
ROD cone dystrophy condition characterized by genetic defects in the production of photoreceptor proteins resulting in bilateral melanin pigment clumping and subsequent early degeneration of rods and of cones in later stages due to a defect in the rhodopsin gene. Presents as loss of peripheral vision with the first symptom usually being loss of night vision. Genetic counseling and referral to low vision indicated. Long term prognosis is poor.
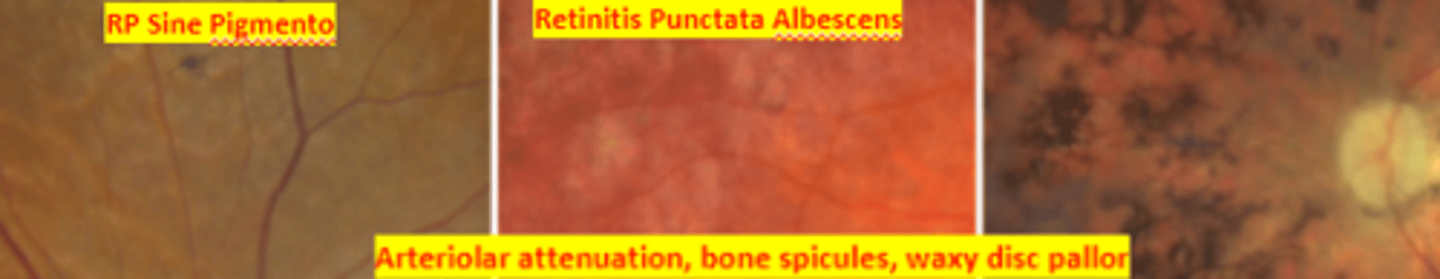
Arteriolar attenuation, retinal bone spicule pigmentation, waxy disc pallor
classic triad of retinitis pigmentosa
Cataracts, myopia, CNVM
three common ocular associations with retinitis pigmentosa
Posterior subcapsular cataract
cataract that is associated with retinitis pigmentosa and can be very debilitating in a patient already having peripheral vision loss
scotopic ERG, pattern ERG, photopic ERG and EOG
Retinitis pigmentosa will show early ____ reduction with normal ____. In later stages _____ will be reduced or absent
Usher syndrome, Freidreich ataxia
two systemic syndromes associated with retinitis pigmentosa
Sporadic
common, non-inherited retinitis pigmentosa caused by a mutated gene that can be passed down to offspring.
Autosomal dominant
inheritance of retinitis pigmentosa that is common with the best prognosis.
Autosomal recessive
inheritance of retinitis pigmentosa that is less common and associated with systemic disorders
X linked recessive
inheritance of retinitis pigmentosa that is the least common and most severe.
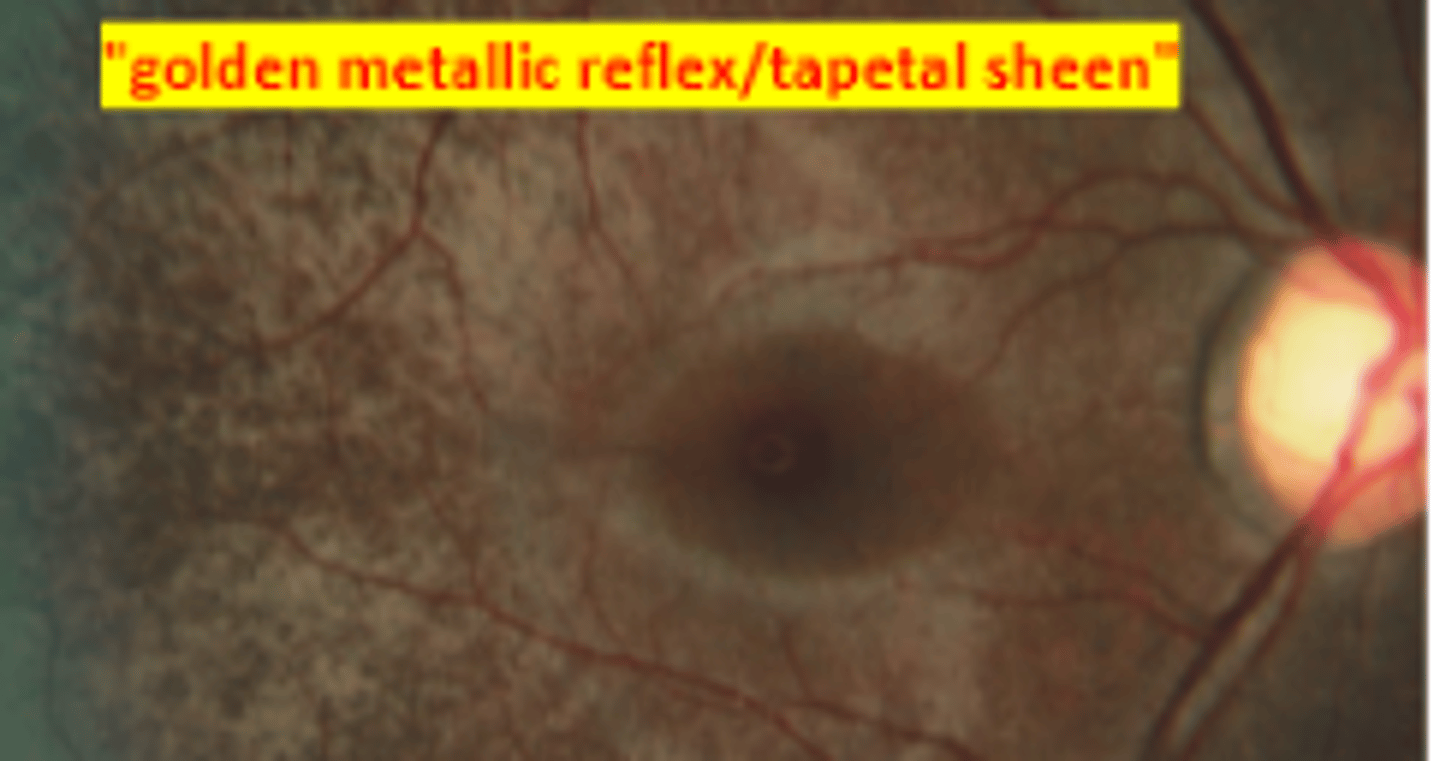
RP sine pigmento
stage of RP where there is arteriolar narrowing, fine dust like intraretinal pigmentation, and loss of RPE
Retinitis punctata albescens
stage of RP where there is scattered white dots most numerous at the equator
Sector
atypical RP occurring only in one quadrant or only in one half (usually inferior) of the retina. Progression is slow and may remain stationary
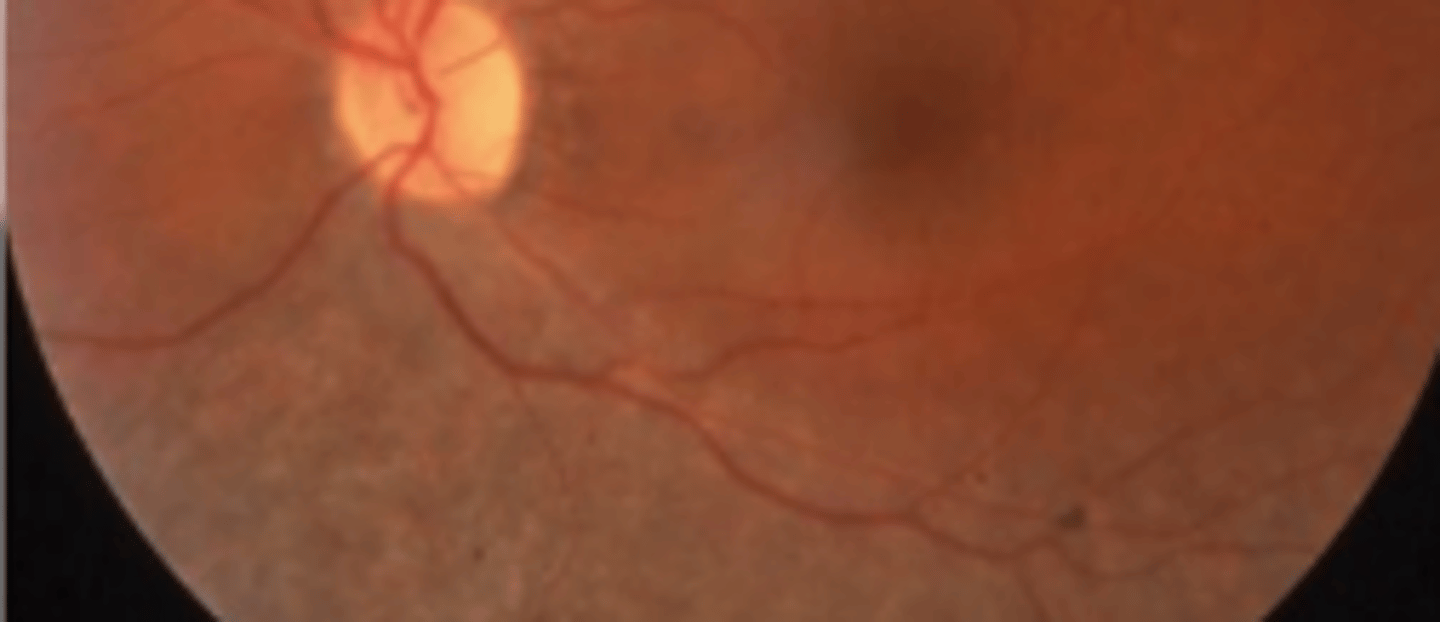
Pericentral
atypical RP emanating from the disc and extending along the arcades
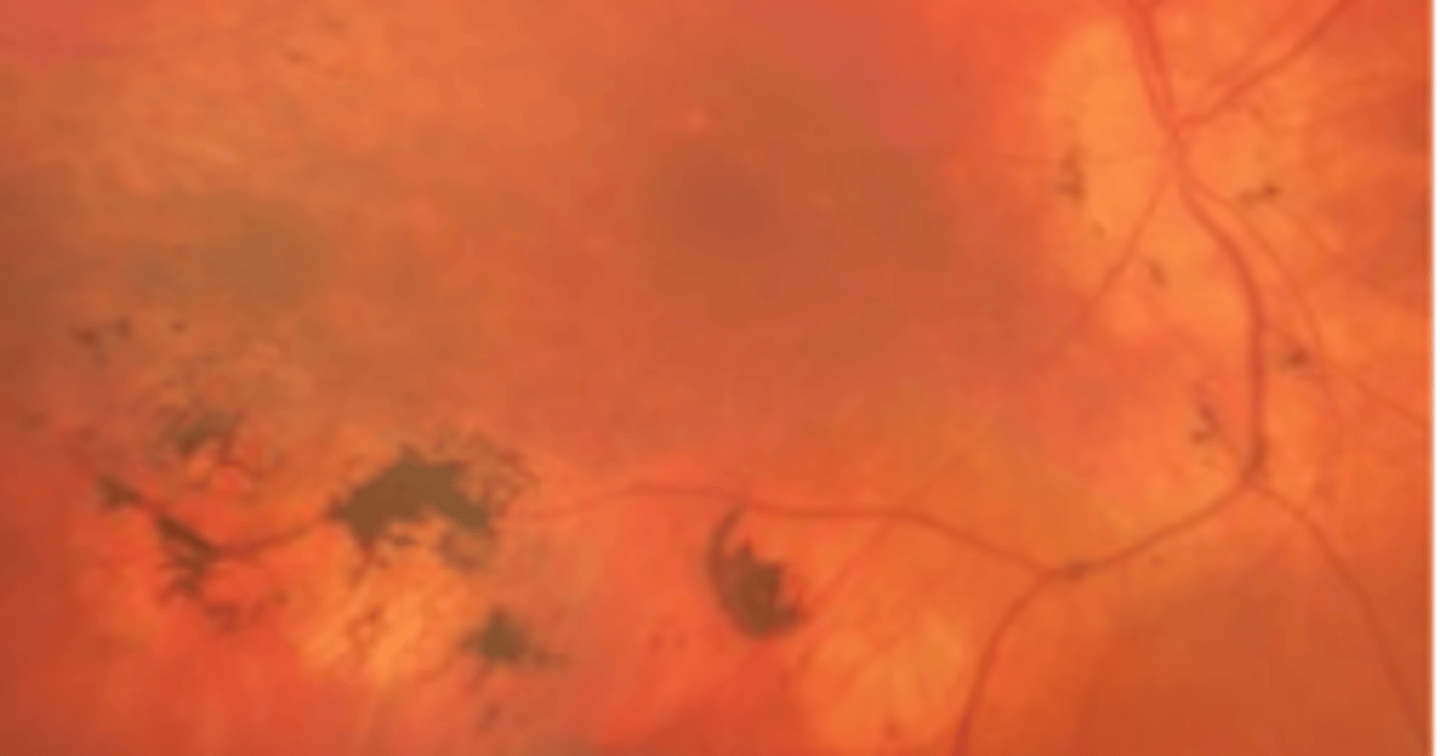
Coats disease
Retinitis pigmentosa with exudative vasculopathy is very rare and appears similar to _____ having lipid deposition in the peripheral retina and risk of exudative retinal detachment.
Luxturna
FDA approved gene therapy used in the treatment of Leber congenital amaurosis and x-linked RP. This drug when injected subretinally enables retinal cells to make RPE65 protein allowing for the visual cycle to continue. Single injection does not cure the disease, but slows progression.
vitamin A
High doses of ____ may slow the progression of retinitis pigmentosa, but this is not currently supported by much evidence and is controversia
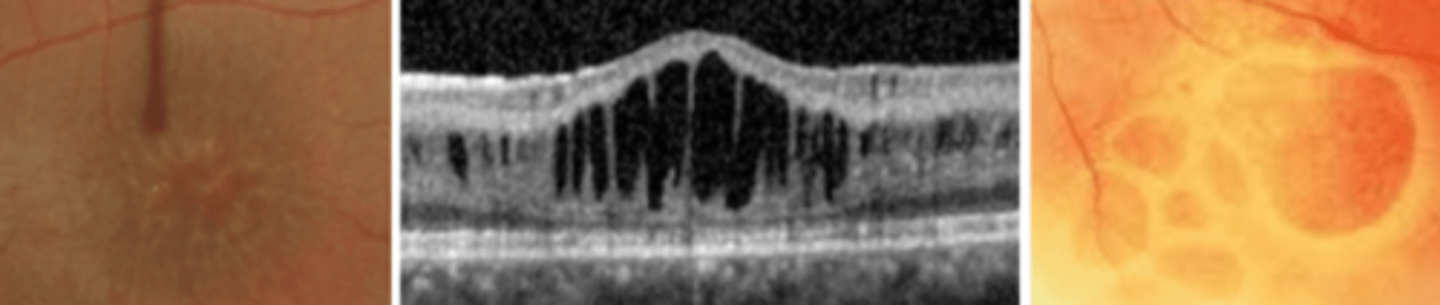
Stargardt Disease (juvenile macular dystrophy)
CONE rod dystrophy which is an autosomal recessive disease causing a mutation of the ABCA4 gene. Presents as bilateral gradual impairment of central vision in the 1st to 2nd decade which may be out of proportion to macular changes which may be appointed to malingering since patients are young. Poor prognosis with no treatment. Genetic counseling and low vision devices are available.

Photopic ERG, pattern ERG
two electrodiagnostic tests that are useful in the early diagnosis of Stargardt disease
Kollner's rule
Upon color vision testing Stargardt disease is an exception to...
Thickened ELM, EZ line absent at the fovea
two findings of Stargardt disease seen on OCT
Fundus Flavimaculatus
an autosomal recessive disease that is considered Stargardt disease presenting in adulthood in the absence of macular involvement. Presents as pisciform yellow white flecks scattered throughout the posterior pole. Good prognosis due to later onset and minimal maculopathy. Geographic atrophy rarely occurs.
Leber congenital amaurosis (LCA)
a group of autosomal recessive inherited retinal dystrophies which is the most common genetic cause of visual impairment in children. Better prognosis if it is the RPE65 gene involved due to the fact that there is gene therapy available. Appears as diffuse white spots of the retina, vessel attenuation, optic nerve edema and pallor. Also presents as nyctalopia, nystagmus, and oculodigital syndrome.
Strabismus
Nystagmus
Hyperopia
Keratoconus
Cataract
5 Ocular Associations of leber congenital amaurosis
Intellectual disability
Deafness
CNS and renal abnormalities
Skeletal malformation
Endocrine dysfunction
Cardiomyopathy
6 Systemic Associations of leber congenital amaurosis
Congenital stationary night blindness
group of bilateral, non-progressive disorders with reduced night vision but normal daylight vision. Can occur with a normal or abnormal fundus.
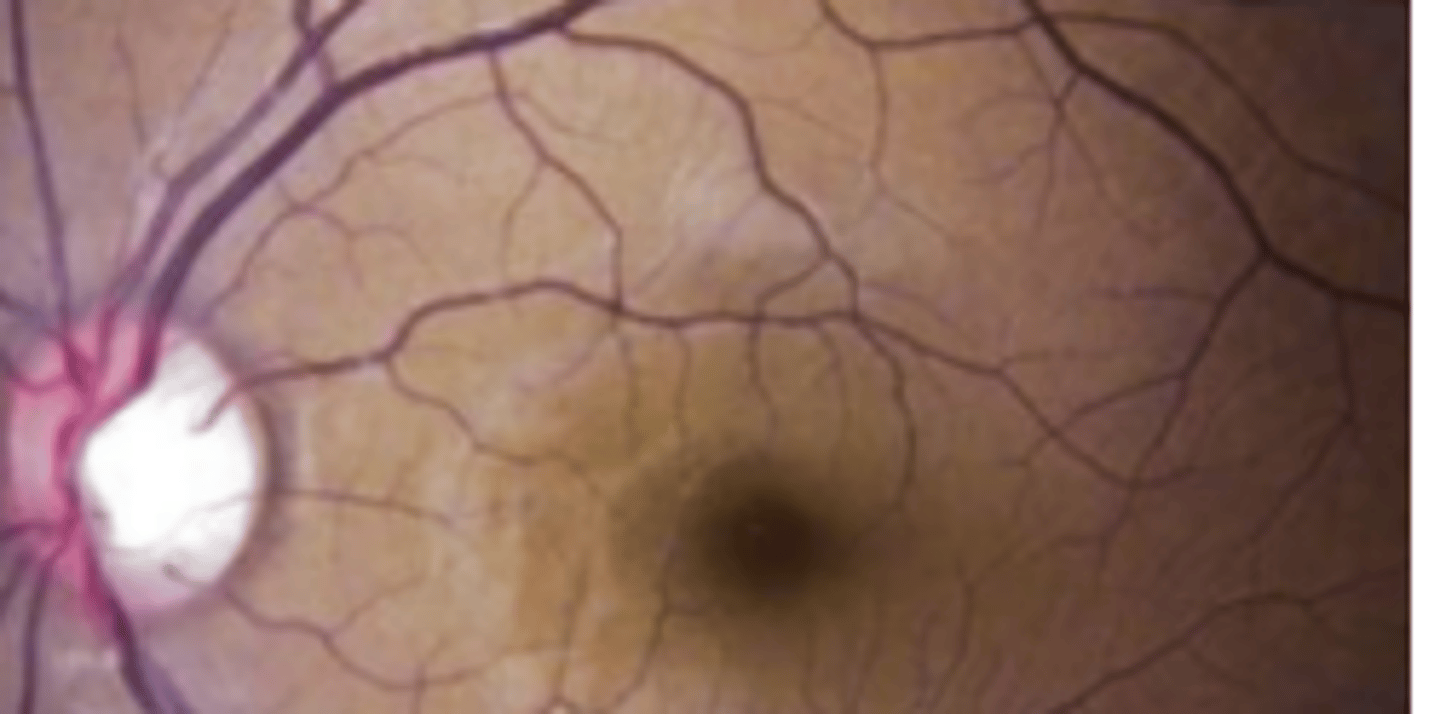
Oguchi disease
autosomal recessive congenital stationary night blindness having a 2-12 hours delay in dark adaptation. The fundus appears golden brown when light adapted and normal colored when dark adapted (Mizuo phenomenon).
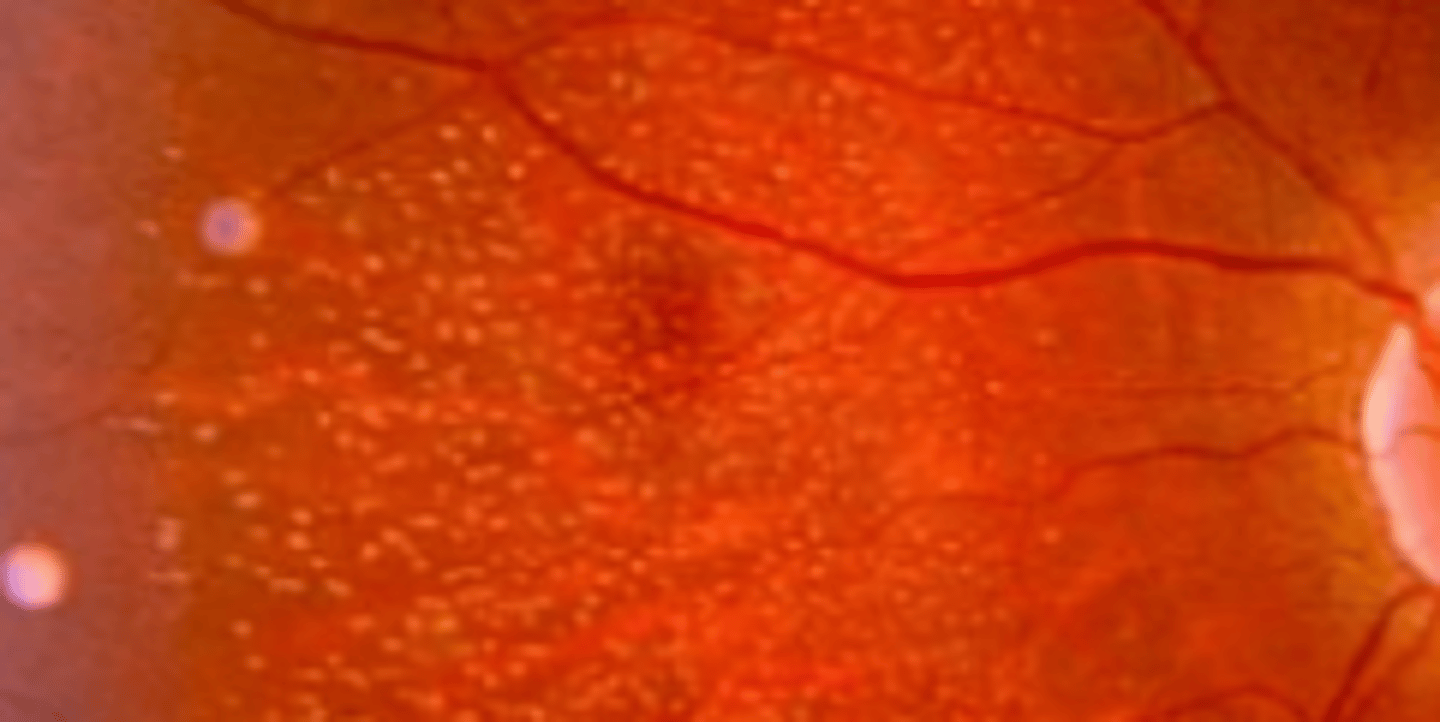
Fundus albipunctatus
autosomal recessive congenital stationary night blindness having tiny yellow spots at the posterior pole that spares the fovea.
Negative B wave
Congenital stationary night blindness shows this characteristic finding on ERG
Congenital retinoschisis
x-linked bilateral maculopathy associated with peripheral retinoschisis due to a defect in Muller cells splitting the NFL from the sensory retina occurring at the OPL. Presents 5-10 years being slowly progressive. Secondary changes include oval defects of the ILM that coalesce and leave free floating blood vessels in the vitreous. Parent education and referral to retina.
Strabismus, nystagmus
two associated ocular findings in congenital retinoschisis that differentiates it from acquired retinoschisis
Vitreous heme, retinal detachment, intra-schisis heme
three complications that may occur even due to minor trauma in patients having congenital retinoschisis
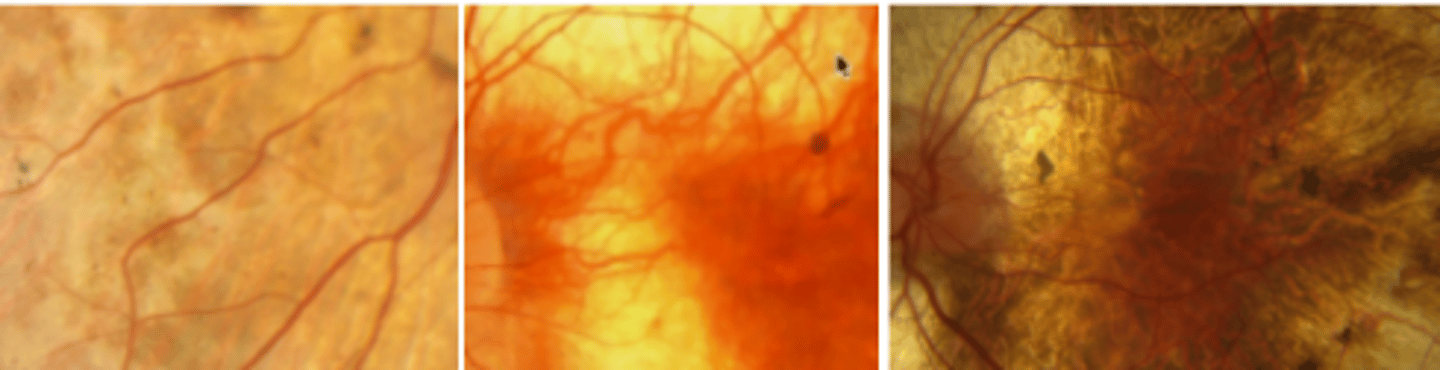
Choroideremia
an X linked progressive diffuse degeneration of the choroid, RPE, and retinal photoreceptors. Presents in the 2nd-3rd decade with nyctalopia and constricted visual fields. Central vision is spared up until the 6th decade, prognosis is poor. ERG and EOG subnormal. VF shows annular scotomas that eventually involve fixation.
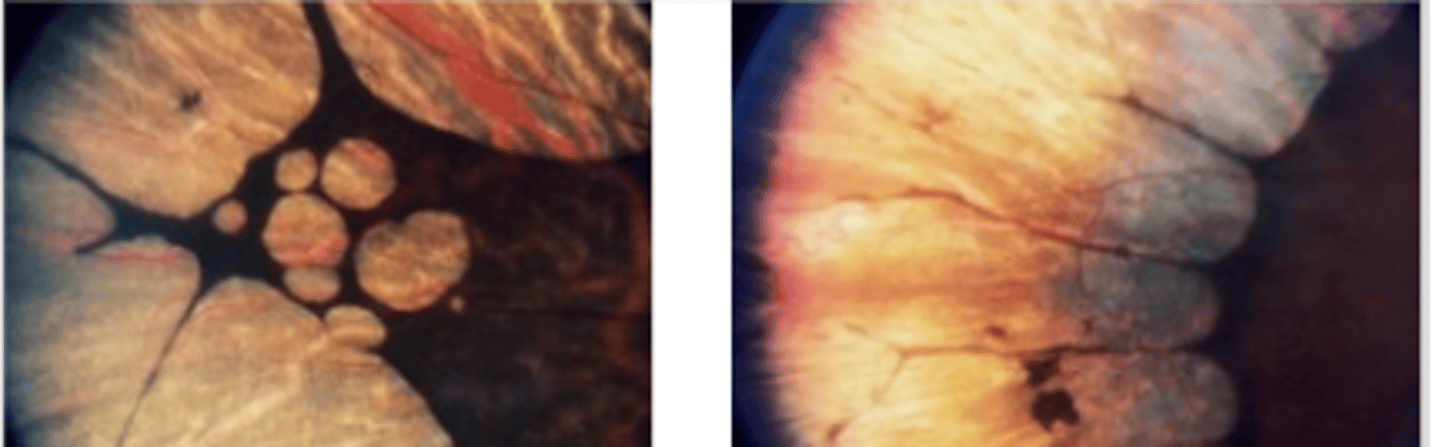
Gyrate atrophy
an autosomal recessive condition resulting in a metabolic defect due to mutation of the ornithine degradation enzyme. Leads to a buildup of ornithine in plasma, urine, CSF, and aqueous. Presents as a reduction in peripheral and night vision. Patches of choroiretinal atrophy coalesces to form scalloped lesions. Spares the fovea until late disease (4th-5th decade), prognosis is poor. Treatment includes pyridozine (vitamin B6) and limitation of arginine intake (low protein).
Central areolar choroidal dystrophy (CACD)
an autosomal dominant condition resulting in progressive significant vision loss due to a mutation in the peripherin 2 (PRPH 2) gene. Progresses in stages, but appears extremely similar to AMD throughout making it difficult to diagnose. Presents as decreased vision and scotoma. Low vision referral and genetic testing indicated.
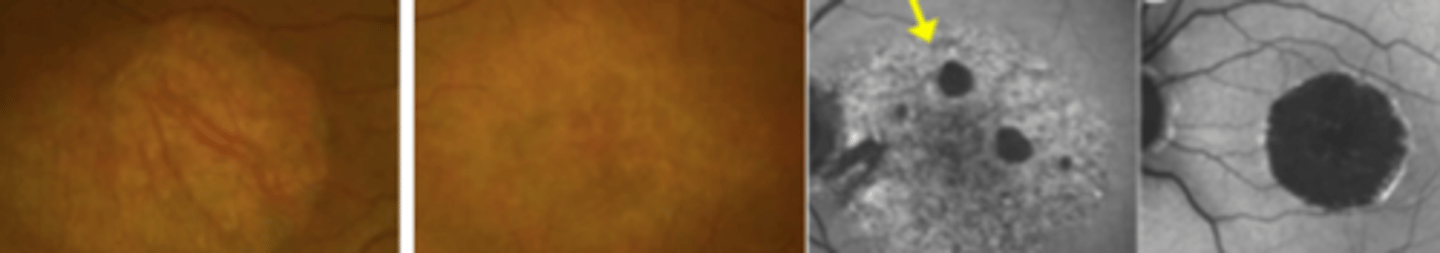
Younger presentation (3rd-4th decade), sharp decrease in vision (5th-7th decade), no drusen
three factors differentiating central areolar choroidal dystrophy from AMD
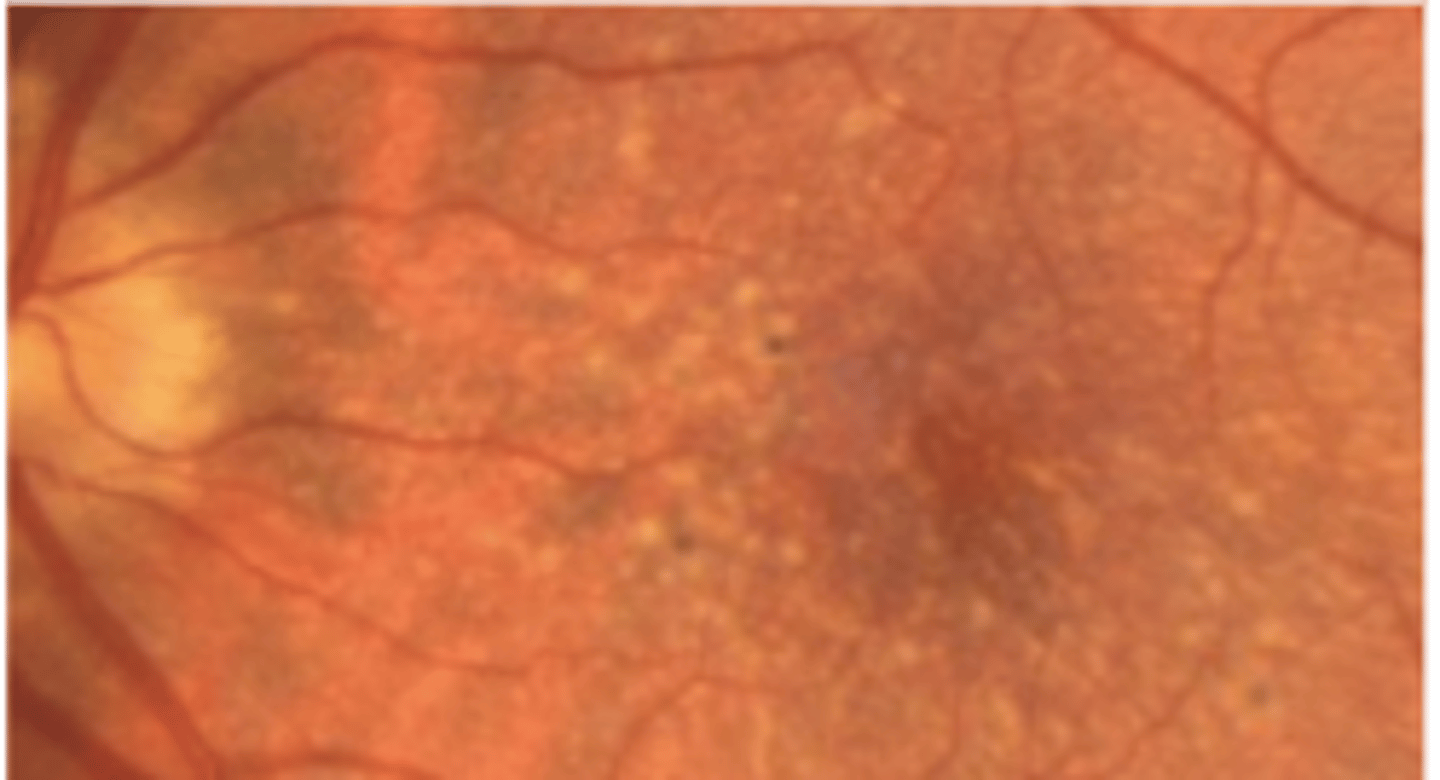
Familial dominant drusen (malattia leventinese, Doyne honeycomb retinal dystrophy)
an autosomal dominant condition presenting as early as 15 yo, but typically in the 2nd-3rd decade. Is characterized by drusen scattered throughout the posterior pole which may result in RPE degeneration, macular atrophy, or CNVM. If macular involvment presents as scotomas, metamorphopsia, and decreased VA.
Albinism
a heterogenous group of disorders affecting melanin synthesis of the eyes, skin, and hair. Vary greater in presentation and prognosis.
autosomal recessive, X linked
Both oculocutaneous and ocular albinism may be _____, but ocular albinism can also be _____
Tyrosinase negative (type I, complete)
albinism with a complete absence of melanin production. Presents with severely decreased vision due to foveal hypoplasia, nystagmus, translucent iris, hyperopic refractive error, and strabismus.
Tyrosinage positive (type II, incomplete)
albinism where there is synthesis of variable amounts of melanin. Pigmentation is variable.
Foveal hypoplasia
absence of the foveal pit. Is the biggest contributor to poor vision in both type I and type II albinism.
Alport syndrome
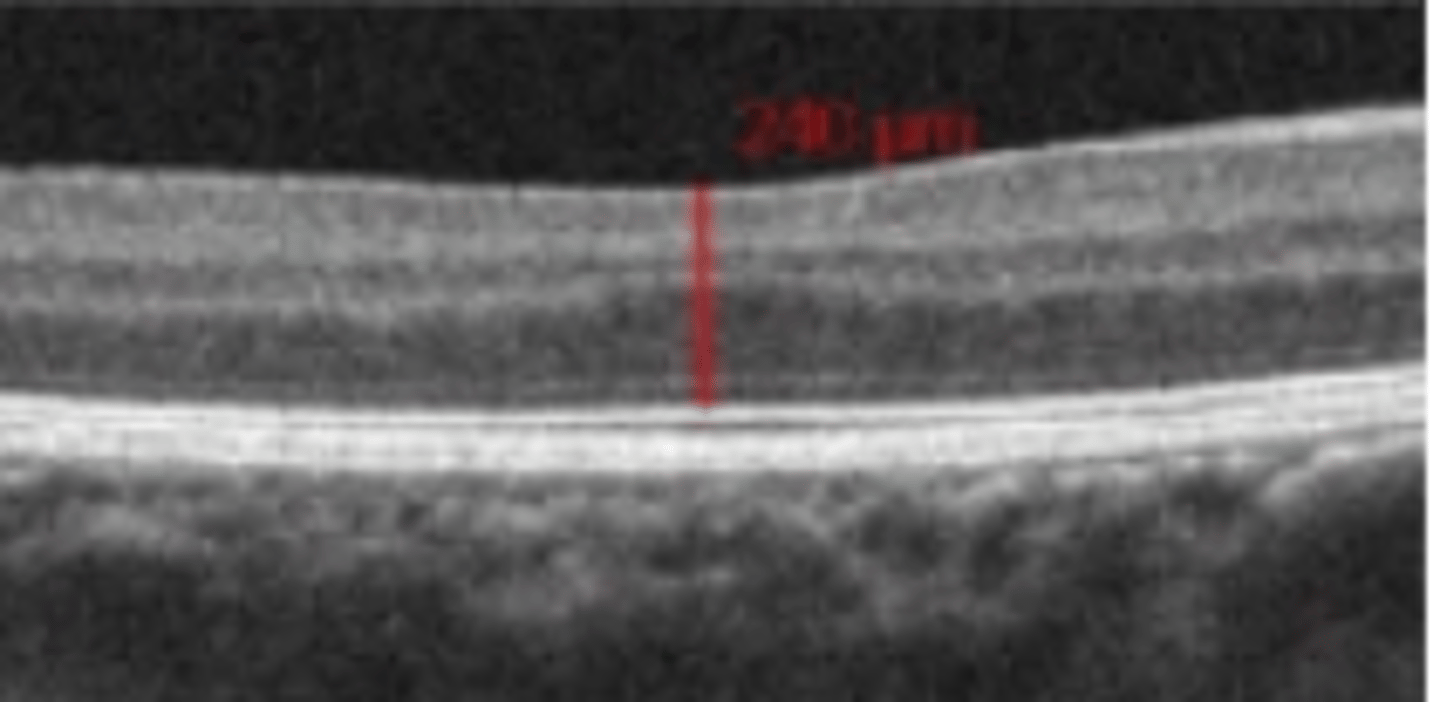
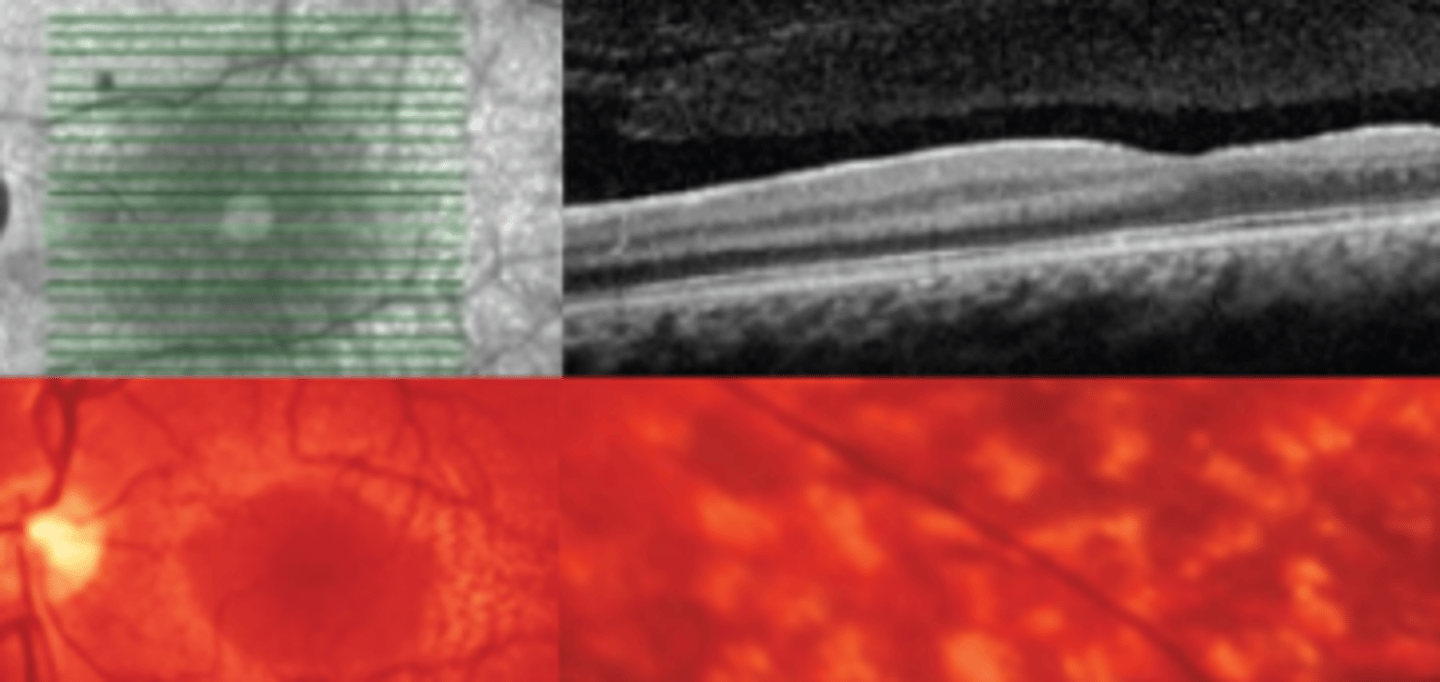
syndrome caused by a defect in collagen type IV synthesis usually presenting in the 2nd-4th decade. Involves kidney failure and sensorineural deafness. Ocular signs include yellow flecks in the perimacular area and temporal macular thinning both sparing the fovea. Also presents with anterior lenticonus and posterior polymorphous corneal dystrophy.
Stickler syndrome
an autosomal dominant syndrome which is a disorder of collagen resulting in abnormal vitreous, myopia, orofacial abnormalities, and deafness. The most common inherited cause of retinal detachment in children. Involves optically empty vitreous, lattice like degeneration posteriorly, high myopia, and lamellar cataract.
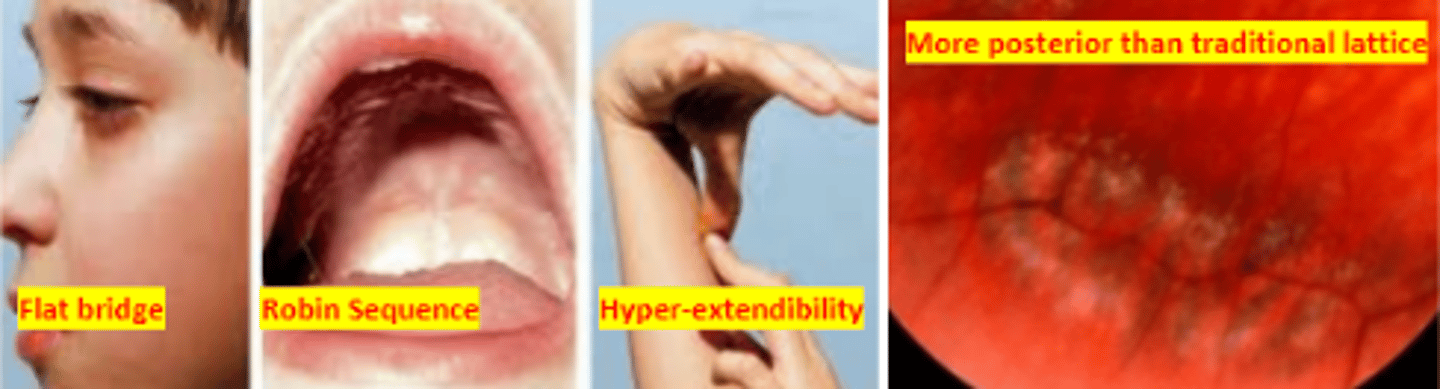
Type I
form of stickler syndrome making up 60% of cases. Has both ocular and systemic features
Type II
form of stickler syndrome involving congenital non-progressive high myopia and sensory neural deafness
Type III
form of stickler syndrome having typical systemic features, but no ocular features.
Confirm diagnosis
Tailoring management plans
Evaluation of family members
Family planning
4 Benefits of Genetic Testing