Clinical immunology exam revision
1/112
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
113 Terms
Why does immuno-regulation occur
To conserve energy
To repair tissue
To prevent damage to non-diseased tissue
The 3 of immuno-regulation
Cell intrinisc mechanisms
Immuno-regulatory molecules and cytokines
Immuno-regulatory cells
Cell intrinisic immuno-regulation
Molecular regulators within cells can:
Turn down signaling pathways after they’ve been triggered such as PRRs
Can turn of chronic/strong signals (exhaustion)
Can limit how many times a cell can divide
Can limit the cells lifespan
Immuno-regulatory cytokine - Interleukin 10, production and function
Production:
Made by many monocytes and lymphocytes
Made later in the immune response
Function:
Suppresses T-cell activation
Promotes class switching and memory cells/LLPC’s
Immuno-regulatory cytokine - Transforming growth factor beta, production and function
Produciton:
Made by macrophages later in the immune response
Function:
Suppresses T and B cell acitvation
Promotes generation of induced T regulatory cells
Promotes stem cell activation and tissue repair
Immuno-regulatory cells - CD4 T regulatroy cell, function and types
Function:
Produces IL-10, TGF-b and CTLA-3
Suppress all macrophages, DCs, T and B cells
Consumes IL-2 (important for immune response)
Types:
Natural = Differentiates in the thymus from cells that are slightly reactive for self-antigen
Induced = Differentiates in the periphery from CD4 T cells activated in the presence of TGF-B or less antigens
Chemokine transportation
To lymphnode = CCR7 → CCL19 & 21
To inflamed tissue (T-cell) = CXCR4 → CXCL9 & 10
To inflamed tissue (monocyte) = CCR2 → CCL2
To inflamed tissue (neutrophil) = CXCR1 & 2 → IL-8
Symbiotic microbiota types
Mutualistic = both host an microbe benefit
Commensalisitcs = no benefit or harm to either organism
Transmissibility
The ability of a microbe to transmit infection from one host to another
Infectivity
The ability of a microbe to establish an infection
Pathogenicity
The ability of a microbe to cause disease upon infection
Virulence
The measure of damage done by a microbe
Viral infection → features
Intraceulluar
Rapid evolution
Zoonosis
Can be enveloped
Single-stranded or double stranded DNA or RNA
Viral infection → immune evasion
Viral proteins supress IFNs
Can downregulate MHCI
Can mutate their antigens
Viral infection → testing
Antigen testing
Molecular testing for viral genome e.g PCR
Serolgical testing for antibodies
Bacterial infection → features
Prokaryotic intra or extracellular pathogen
Contain many PAMPs
LPS
unmethylated CpG motifs in DNA
Flagellin
Carbohydrate capsule
Can undergoes zoonosis
Can undergoes dormancy/latency
Bacterial infection → immune evasion
Protease expression to degrade antimicrobial chemicals
Bacteria can swap their capsule and other antigens in different life phases
Bacterial infection → testing
Culture/grow bacteria
Microscope analysis for bacteria presence (post staining)
Serological testing for antibodies
Antigen testing
Molecular testing for genome e.g pcr
Parasitic infection → features
Intra or extraceullular eukaryotes
Protozoa = single celled
Helminth = worm
Ectoparasite = live on skin e.g tick
Parasitic infection → immune evasion
Multi-stage lifecycle leads to cycling antigens
Can secrete immuno-suppressive proteins and enzymes
Parasitic infection → testing
Serological testing for antibodies
Molecular test for parasitic genome (PCR)
Microscopic test for parasite presence
Fungal infection → features
Eukaryotic, extracellular infection
Yeast = single-celled organism
Mycelial = multi-celled organisms
Fungal infection → testing
Culturing/growing fungus
Microscopic test for fungal invasion
Serologic testing for antibodies
Immunological memory → requirements and function
Requirements:
Innate immune response
Antigen uptake and presentation
Adaptive immune response
Function:
Increases frequency of memory B or T cells
Decreases the amount of signal needed to activate memory cells
Passive immunisation → what, when to give, protection duration and good for
Transfer of antibodies from an immune to non-immune organism
Ideally given before infection
Immediate protection with limited duration
Good for toxins and outbreaks where vaccines are not developed
Active immunisation → what, when to give, protection duration, 4 main outcomes
Induction of a specific, protective immune response
Must be given well in advance as it take weeks to develop immunity
Long duration i.e. years
Outcomes include:
Eradication
Sterilizing immunity
Reduce disease symptoms
Reduce infection transmission (Herd immunity)
Herd immunity
When immunity protects the individual and benefits the broader community
Herd immunity threshold (HIT) → description and dependancies
HIT is the frequency of people in a population that need to be immune to prevent widespread transimssion
Dependant on:
Infectiousness of the pathogen (R0)
Frequency of:
Infected individuals (resevoir)
Vaccinated individuals who dont develop protective immunity (vaccine efficacy)
Individuals who arent vaccinated (vaccine coverage)
Infected/vaccinatde individuals who develop protective immunnity
2 main components of a vaccine
Adjuvant = A compound that augment and/or enhance the immune response
Antigen = A component of the pathogen that is immunogenic
Criteria for selecting suitable antigens
1: Abundantly expressed and accessible to protective immune mechanisms e.g antigen expressed on cell if T-cells needed or antigen expressed on pathogen if B-cells are need
2: Does not vary during a pathogens life cycle
Adjuvant mechanism of actions
Augment the immune response by:
Trigger innate PRRs via PAMPs
Each PRR triggers a differnt set of cytokines that acts differently on the adaptive immune system
Promote antiegn presentation by DCs
Increase cytokine production
Directly promote uptake of antigen by DCs
Clinical testing phases

Types of vaccines

Hypersensitivity and examples
An exaggerated immune response directed against an antigen that is harmless e.g allergy, autoimmunity and transplantation
Types of antigens that trigger hypersensitivity
Allergen = antigen from environment
Auto-antigen = self antigen
Allo-antigen = non-self antigen from transplanted tissues
Type 1 hypersensitivity → Phases and associated granular contents
Phase 1 (sensitisation) = B-cells present allergen to CD4 T cells which produce cytokines IL-4,5 and 13 which promote B-cell activation and class switching to produce IgE which load onto mast cells and basophills
Phase 2 (activation) = Allergen crosslinks IgE on the surface of mast cells/basophils which triggers degranulation
Granular contents include:
Histamines
Leukotrienes and prostaglandins
Cytokines/chemokines
Type 2 hypersensivitiy → phases
Phase 1 (sensitisation) = B-cells take up the antigen and either activate with a t cell (IgG) or without (IgM) into plasma cells which secrete the antibodies into circulation
Phase 2 (activation) = Complement proteins, phagocytes or NK cells encounter the antibody bound to the antigen and become activated
Type 2 hypersensitivity → description, immune cells/antibodies and causes
Antibody-mediated cytotoxic hypersensivitiy that builds when IgG or IgM in circulation bind to antigens on the surface of fixed tissues and trigger complement activation, phagocytosis or NK cell activation against the tissue
IgG or IgM + complement, phagocytotic, or NK cells
Caused by a failure of tolerance (self-antigens triggerring autoantibodies) or exposure to transplanted tissues (antibodies directed against allo-antibodies)
Type 1 hypersensitivity → description, immune cells/antibodies and causes
Immediate hypersensitivity that builds when IgE antibodies bind to a multivalent allergen and trigger a response in granulocytes
Involves IgE + Mast cells, basophills and eosinophils
Caused by an imbalance between Th1 and Th2 (more Th2 = more humoral) due to:
Air pollution
Genetics
High income
Low microbiota
Type 1 hypersensitivity symptoms → phases and examples
Early = within minutes, mediated by mast cell and basophill degranulation
Late = within hours, mediated by eosinophills and neutrophils
Symptoms:
Itchiness
Swelling
Mucus production
Breathing problems
Anaphylaxis (systematic)
Type 2 hypersensivity testing (indirect and direct)
Indirect = testing for preperation of blood transfer
Mix patient plasma with donors blood and anti-human globulin (coombs reagent) to check for agglutination
Direct = testing to diagnose after blood transfer
Mix patient blood with coombs reagent to check for agglutination
Type 1 hypersensitivity treatment (local, systematic and desensitiisation) and examples
Local = block immune mechanisms
Anti-histamines block H1 receptors and block histamines
Leukotriene antagonists to block leukotrienes
Inhibitory steroids generally
Systematic = epinephrine prevents airway constriction
Desensitisation = repeated low dose exposure to allergen may reduce T1 hypersensivity
T1 hypersensivitiy testing
Apply a pael of allergens to skin, wait 15-20 mins, positive = wheal (raised tissue) and flare (redness), negative = nothing
Type 3 hypersensitivity → description, antibody/cells involved, physical causes
High concentrations of IgG or IgM antibodes form immune complexes that bind to blood vessel walls/deposit in tissues to cause tissue damage, complement activation and neutrophilr recruitment
Antigen/antibody complexes in tissue/circulation + complement and neutrophills
Causes:
Self-antigen (tolerance failure)
Insect bite
Transplanted tissues
Repeated administration of foreign protein e.g vaccinating whilst already having antibodies
Type 3 hypersensitivity → phases
Phase 1 (sensitisation) = B-cells take up antigen and either activate by themselves (IgM) or with CD4 T cells (IgG) into plasma cells which produce high levels of antibodies
Phase 2 (activation) = Antigen binds to antibodies in circulation/tissue and form immune complexes that deposit in blood vessels/tissues and trigger strong local immune responses
Type 3 hypersensitivity → symptoms and testing
Arthus reaction
Damage to BV can cause leakiness, swelling and bruising around site
Testing:
staining tissue sections to look for immune complexes
Testing plasma for:
Complement levels (decreasesd if high disease activity)
Anti-rheumatoid factor (RA)
Anti-nuclear antibodies (lupus)
Type 4 hypersensitivity → description
Delayed hypersensitivy as a result of sensitised cd4 t cells and macrophages and infections or allergens
Type 4 hypersensitivity → phases
Phase 1 (sensitisation) = DCs present antigens to CD4 T cells which become activated and prolifereate into TH1 cells
Phase 2 (activation) = DCs activate memory Th1 CD4 cells which migrate to site of inflamation and produce Interferon gamam to recruit and activate macrophages
Phase 2.5 (prolonged effector phase) = Occurs if macrophages cannot clear the antigen, triggering chronic inflammation, fibrosis and granuloma
Type 4 hypersensitivity → testing
Inject an antigen intradermally and wait 48-72 hours for a wheal (raised skin) and flare (redness) reaction
Central vs peripheral tolerance
Central = negative selection in the thymus and bone marrow
Peripheral tolerance = regulatory cells such as
Natural Treg cells (self-Ag)
Induced Treg cells (low Ag)
Anergy (Ag with no co-stim)
Hashimoto’s disease → hypersensitivity types, symptoms and testing
Autoantibodies (type 2) and T-cell sensitisation (type 4) to thyroid antigens
Symptoms include goiter (swollen thyroid/neck) and T4/3 deficiency
Testing incldues looking for anti-thyroid peroxidase,thyrogloublin antibodies or low T4 yet high TSH
Type 1 diabetes → hypersensitivity types, symptoms and testing
Autoantibodies (type 2) and T cell hypersensitisation (type 4) to insulin producing beta cells in pancreas
Symptoms include hyperglycaemia, nerve and kidney damage
Testing includes looking for anti-glutamic acid decarboxylase antibodies, high blood glucose and glycated hemoglobin
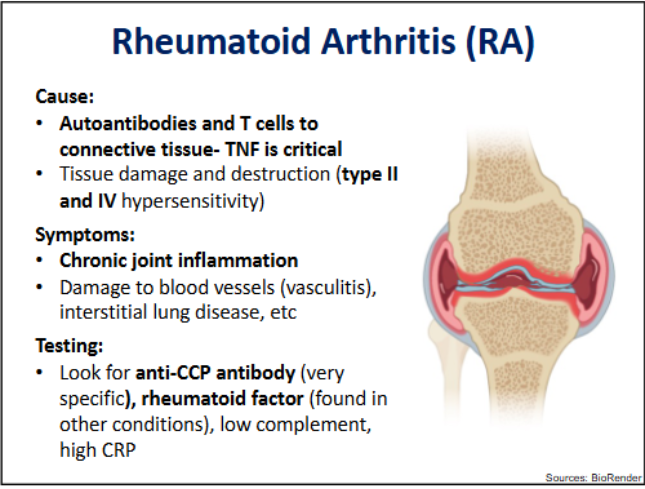
Rheumatoid Arthiritis → hypersensitivity types, symptoms and testing
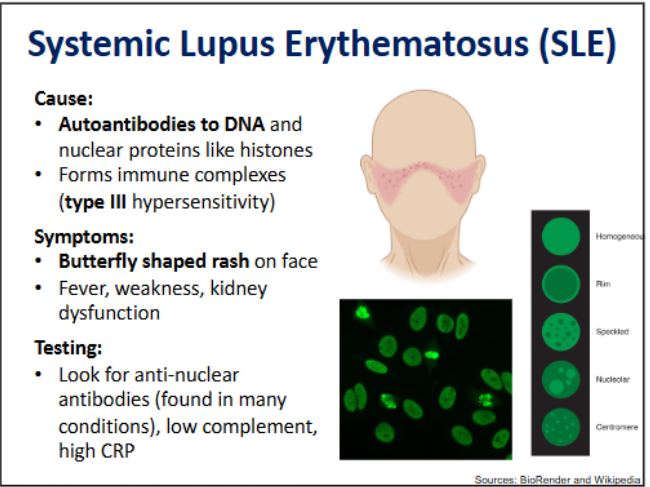
Systematic Lupus Erythematosus → hypersensitivity types, symptoms and testing
Types of tissue grafting
Autograft = from self
Isograft = from identical twin
Allograft = from a member of the same species who is not genetically identical
Xenograft = from other species
Commonly transplanted tissues
Blood transfer which includes:
RBC’s platelets and plasma
Must deplete donor WBC to prevent graft vs host disease
Can be done through size exclusion filters during aphresis (how we get blood out of individiuals) or gamma irradiation
Organ transplants which includes:
Heart, liver, lung and kidney
Bone marrow transplants, haematopoetic stem cells or umbilical cord blood
To replenish WBCs
Skin
Like a bandage
Autoimmunity → Major histocompatibility antigens/human leukocyte antigen class 1 v 2
The most common antigen known to trigger autoimmunity after organ transplants.
Class II has 2 chains:
heavy chain can be encoded for by 3 possible genes (HLA-A,B or C)
beta-2-microglobulin
Class II has 2 chain:
alpha chain can be encoded for by HLA-DP, DQ or DR genes
Beta chain can be encoded by 3 different genes of the same name (generally pair with alpha chain)
Depending on parents and homo/heteozygousity humans can express typically 6 MHC I complexes and 6-8 MHCII complexes on 1 cell
Minor histocompatibility antigens (MiHA)
Normal proteins that are polymorphic, the differences between humans can be a less frequent cause of graft rejection
Mostly coded on y chromasome
Outcomes of MHC/MiHA mismatch
Transplant services aim to match donor recipient with most polumorphisms however less matching can still result in high degrees of survival due to high dose immunosuppressants stem cells excluded
Xenograft matching
Cannot match MHC or MiHA
Certain proteins are expressed in different species but not humans
Rhesus D antigen, pregnancy and preventative measures
Ion channal proteins on the surface of RBCs, the most important one being the D antigen:
RH D negative = can recieve Rh D transfusion once, after which IgG generation against Rh D antigen occurs
Pattern occurs in pregnancy with Rh D- mother and Rh D+ father, causing haemolysis in the sercond Rh D+ fetus
Anti-D IgM prevents this by mopping up fetal RBC’s before they can enter the bloodstream but fails to cross the placenta
A and B RBC/platelet antigens → description, and allelic variants
Carbohydrate modifications on the surface of RBC’s, there are 2 dominant allelic variants (A, B) and 1 submissive null variant (O)
Blood type A can recieve A and O blood
Blood type B can recieve B and O blood
Blood type AB can recieve A, B. AB and O blood
Blood type O can recieve O blood
Plasma transfusion
The inverse of blood transfusion
Determining blood/plasma type
Mix patient blood/plasma with reference blood/plasma and coombs reagent and look for agglutination
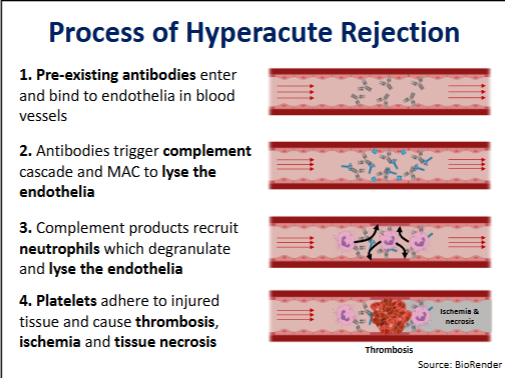
Hyperacute rejection → timespan, symptoms, causes, treatment
Timespan = minutes to 48 hours
Symptoms = thrombosis (blood clot), followed by ischrmia (loss of blood flow) and necrosis (death of tissue)
Causes = pre-exisiting antibodies in the recipient graft - T2 hypersensitivity
Treatment = remove tissue
Hyperacute rejection management
Prior to transplant:
Screen for pre-existing antibodies
e.g transplant history or complement dependent cytotoxicity crossmatch assay
After transplant
Monitor tissue and patient
Hyperacute rejection process
Acute rejection → timespan, symptoms, causes, treatment
Timespain = days to weeks after transplant
Symptoms include:
T cells mediated interstitial lymphocytic infiltration and focal necrosis of tissues
B cell mediated vasculitis, thrombosis, ischemia and necrosis of vasculature
Causes = driven by new T cells or antibodiies against recipient MHC or MiHA (T2 and 4 sensitivity)
Treatment = active management via depletion of T cells
Process of acute rejection → indirect vs direct
Both involve DCs taking up graft Ag in graft tissue and presenting it to T cells in the lymph node.
Direct = donor DC present their non-self MHC + tissue
More potent in allograft
Weak in xenograft due to MHC being biologically incompatible in edition to other mismatches (e.g incorret costimulatory molecules)
Indirect = recipient DC presents tissue (only dependent on antigen)
Weak in allograft
Potent in xenograft due to being able to present all of the antigens
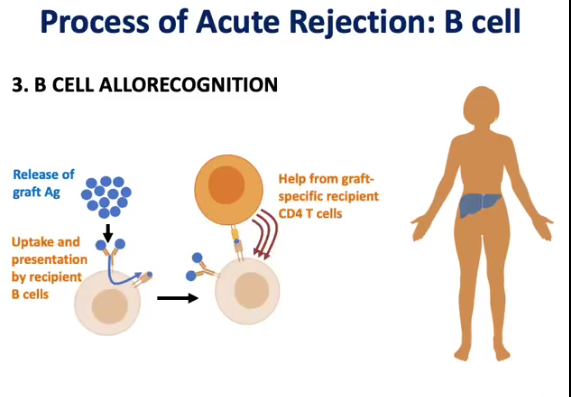
Process of acute rejection → b-cell
Acute rejection management
Prior to transplant
Screen for matching MHC loci
e.g serological or molecular matching
After transplant = immunosuppression
Post-trasnplantation immunosuppression - broad therapy
Conventionally there are 3 different targets of immune therapy (triple therapy)
Inhibition of T-cell activation
Tacrolimus blocks activation and IL-2
Block proliferation
Azathoprine as a purine analogue disrupts cell division
Block inflammation
Corticosteroids inhibit transcription of inflammatory genes
Post-transplantation immunosuppression - targetted therapy
Involves the useage of antibodies and fusion proteins
Antibodies can deplete lymphocytes from graft e.g
Anti-thymocyte globulin depletes T cells
Rabbit-derived polyclonal serum specific for human leukocytes
Inhibit T cell activtion e.g
Anti-CD3 mAb
Anti-IL-2 mAb
Chronic graft rejection → timespan, symptoms, causes, treatment
Timespan = months to years after transplant
Symptoms = fibrosis in intersitial tissues, arteriosclerosis (thickened intima due to proliferation of endothelia & SM), ischemia and atrophy
Cause = T cells generated by the recipient against the graft and graft damage that releses more antigen (acute rejection episodes)
Management of chronic rejection
Before transplant:
Screen for matching MHC and MiHA loci
Siblings tend to perform better than unrelated
After transplant:
Immunosuppression can only delay/reduce chronic rejection
New graft
Graft versus host disease → desription, frequency amongst specific transplantations and treatments
T cells from graft targetting recipient tissue, often the skin, gut and liver
Frequent among bone marrow or stem cell transplantation
Infrequent among transplanted organs
Can be treated with T-cell depletion and immunosuppression
Graft versus host disease → acute vs chronic features
Acute = <100 days:
Skin rash, liver dysfunction and jaundice, nausea and diarrhea
Chronic = >100 days:
Skin, liver and intestinal symptoms as well as other systems e.g vitiligo and alopecia
Management of GvHD
Before:
Deplete bone marrow or stem cells of T cells
MHC match (related individuals help)
Use umbilical cord blood
After:
Prophylactic (preventative) immunosuppression
Additional immunosuppression if it occurs
Cancer/Neoplasm → general description and 3 growth types
When a cell begins to lose control of its cell cycle and grow abnormally, forming a neoplastic growth
3 types of neoplastic growths are:
Benign = unable to invade surrounding tissue or grow indefinetly (can become malignant by accumulating multiple genetic alterations - malignant transformation)
Malignant = start to invade surrounding tissue (cancer)
Metastatic = start to leave primary tumour site and migrate to other distant tissues (mestastision)
Malignant neoplasm types → how and different types
Grouped based on the original cell type
Carcinoma = epithelial origins
Sarcoma = connective tissue origins
hematological cancers:
Leukaemia = less differentiated hematopoeitc origin cells inside the bone marrow
Lymphoma = more differentiated hematopoietic origin cells outside the bone marrow e.g lymph node
Myeloma = plasma cells in bone marrow
Cause of neoplasm and factors that can increase the likelhood
Caused by genetic alterations in the DNA related to the cells division, survical or death
Factors that increase the likelhood of DNA damage is
Chronic inflammation
Carcinogenic chemicals
Ionising radiation
Oncogenic viruses
Genes that cause cancer
Oncogenes = inappropriately expressed genes that promote cell division and survival
E.g Mutated Her2 can upregulate EGF production resulting in excesscell division
Proto-oncogenes = genes that promote normal division and survival, they become oncogenes when dysregulated due to mutation
E.g Her2 typically form the receptor for epidermal growth factor (EGF) which when HER binds to signals cells to divide
Mutated tumour-suppress = inhibited due to mutations, permitting abnormal growth
Tumour-suppressor genes usually prevent inappropriate cell division or survival by promoting cell death or DNA repair
E.g p53 senses ocogenes and DNA damage
DNA mutation → chromosomal translocations and example
Genetic material from one chromasome is swapped with another, there tends to be hotspots where this occurs e.g
End of Chr 8 with Chr 14’s
Shifts the proto-oncogene myc close to an enhancer on Chr 14, converting it to an oncogene
Expressed at high levels in B-cells, leading to Burkitt’s lymphoma
DNA mutations → acquired vs germ-line mutations
Acquired = after birth e.g p53 mutation and infection with oncogenic viruses
Germline = inhertied e.g BRCA1/2 mutations can be inherited and predispose to the development of breast and ovarian cancers
Malignant transformation steps
initiation = genetic alteration that changes cell division or survival (benign)
Promotion = accumulation of cells (benign)
Progression = additional genetic alterations that enable unlimited proliferation and development of malignancy (malignant)
Metastatis = malignant cells lose adhesion and move away from original state (metastasis)
Tumour antigens → 4 types
Overexpressed antigens = normal genes that are overexpressed in tumours (tumour-associated) e.g EGF receptors
Oncofetal antigens = normally expressed only at certain stages of development (tumour-associated)
Neoantigens = mutated versions of normal genes due to carcinogens or radiation (tumour-specific)
Oncoviral antigens = expressed in cells after infection with an oncogenic virus (tumour-specific) e.g HPV protein E6 which inhibits p53
Tumour immunity → cells
NK cells can cells that are stressed, have downregulated MHCI or via antibody dependent cellular cytotoxicity
Macrophages can phagocytose cancer cells or generate cytokines
CD8 T cells can kill malignant cells expressing tumour specific antigens with CD4 T cell assistance - can be tumour infiltrating lymphocytes
B cells can make anti-tumour antibodies (can be problematic by masking from CD8 T cells)
Tumour immunity → cytokines
IL-12 promotes strong CD4 Th1 (cytotoxicity) and CD8 T cell responses
Type I IFN and IFN gamma can enhance tumour immunity
TNF can promote cancer apoptosis
Tumour microenvironment → desription, 4 parts and the impact of chronic inflammation
An environment comprised of the tumour cells and other cells recruited by the tumour using cytokines:
Tissue remodelling from fibroblasts that have been converted to cancer-associated fibroblasts (CAFs) by TGF beta
Incresed invasion via epithelial cancer cells that have been converted to mesenchymal cancer cells by IL’s and TNF A
Angiogenisis (abnormal vessel growth) via VEGF
Immune evasion via the recruitment of supressive myeloid derived suppressor cells from macrophages and T reg cells
Chronic inflammation supports the microenvironment by increasing cellular stress signals (causing the release of the growth factors and cytokines above), increasing mutation rate (genotoxic stress) and it is a proangiogenic
Cold tumour → cells excluded and included, and diagnostic implication
CD8 T cells and NK cells excluded
CD4 Treg cells present
Poor prognosis and response to immune-based therapies
Hot tumour → cells excluded and included, and diagnostic implication
CD8 T cells and NK cells present
CD4 Treg cells excluded
Improved prognosis and response to immune based therapies
Immune editing → description/downside and steps
The process by which cancer cells are eliminated, can lead to a natural selection of only the best cancer cells
Steps include:
Initiation → cancer forms
Elimination → cytotoxic mechanism elmiminate abnormal cells
Equilibrium → a balanced state between cancer regeneration/survival and elimination
Escape → the best (aggressive, less immunogenic) cancer cells survive and create their microenvironment
How cancerous cells evade the immune system
Tumour micro-environment - contains suppressive cells and high antigen load exausts T-cells
Antibody masking - hidden immunogenic epitops on tumour-specific antigens can limit CD8 T cells (antibodies can unintentionally do this)
MHCI down regulation - reduces presentation of tumour-associated antigens which reduces activation of CD8 T cells
Poorly immunogenic tumour antigens are poorly immunogenic/self
Polyclonal vs monoclonal antibodies → location, creator and target
Antibodies in serum are polyclonal
created by B cells
target multiple distinct epitopes
Therapeutic and labelling antibodies are monoclonal
created by cloned b cells
targets a single epitope
Monoclonal antibodies usecases
Labelling e.g tagging her2 breast cancer for identification
Blocking e.g blocking receptor on cancer cell
depleting e.g depleting TNF concentrations in auto-immunity
Activating e.g activating CD3 on T cells
Targetting e.g targeting Her2 cancer cells with conjugated drugs
Monoclonal antibodies production
immunise animals with target antigen
Isolate plasma cells
fuse with specialised tumour cells to form hybridomas (immortal plasma cells)
Clone out a single hybridoma with antibody for target antigen
Humanise antibodies to reduce immunogenicityt
T-cell inhibitory molecules that are upregulated by tumours presence
CTLA-4
Competes with cd28 (costimulation)
Switches T cell activation off
PD-1
Delivers the exhaustion signal to T cell
Immune checkpoint blockade
Monoclonal antibodies bind to PD-1 and CTLA-4 receptors and prevents the molecules from binding to said receptors which would exhaust/inactivate T-cells. also drives said molecules to interact with cd28 instead enabling costimulation
Car T cell therapy → prepeartaion and attack
Living drug for lymphoma that uses modified patient immune cells that express synthetic tumour speciic receptors (Chimeric antigen receptor)
Preperation (3 weeks)
Isolate and activate T cells, transduce them with DNA that encodes tumour-specific synthetic receptor, expand, isolate and reinfuse
Attack (weeks to months)
Migrate around the body, encounter tumour cells with CAR and activate
CAR description and advantage and what makes a good one
Uses short chain variable fragments attatched to a hinge domain and CD3 domain (sends activation signal) and CD28 domain (costimulation domain)
Can bind to membrane bound unprocessed proteins, doesnt need mhc, antigen processing or presentatino
A good CAR must be:
Highly expressed
accessible on cancer cell surface
Unlikely to be mutated