Protein Aggregation
1/14
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
15 Terms
Competing Pathways: Folding vs Aggregation
some proteins can take off-pathway routes, leading to misfolding and aggregation
exposed hydrophobic regions and β-sheet structures in partially folded or misfolded intermediates can non-specific interactions leading to promote aggregation
aggregates typically occupy low-energy states in the protein energy landscape (highly stable)
molecular chaperones avert aggregation by shielding hydrophobic residues and preventing kinetically stable non-functional structures (misfolded intermediates)
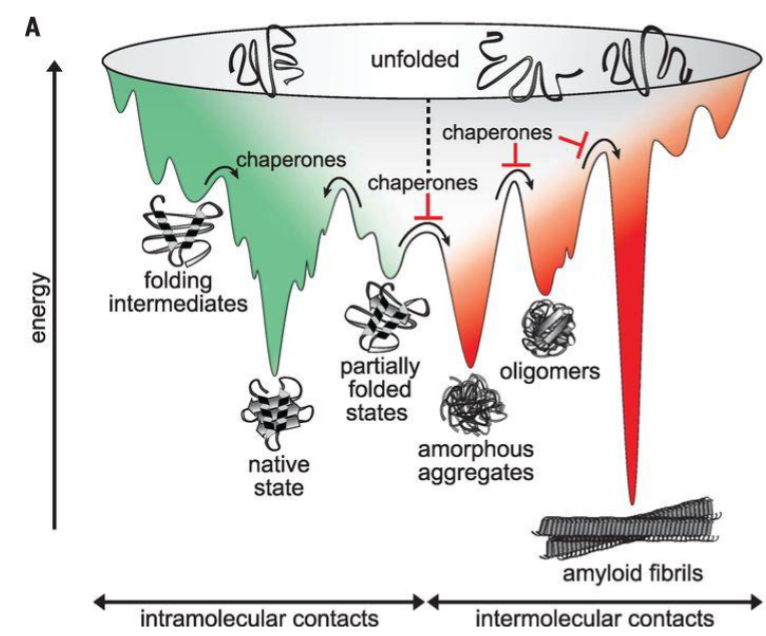
Proteostasis
proteostasis = protein homeostasis
the protein homeostasis network encompasses a large number of proteins that maintain the proteome functional by directing protein synthesis, folding, trafficking and degradation
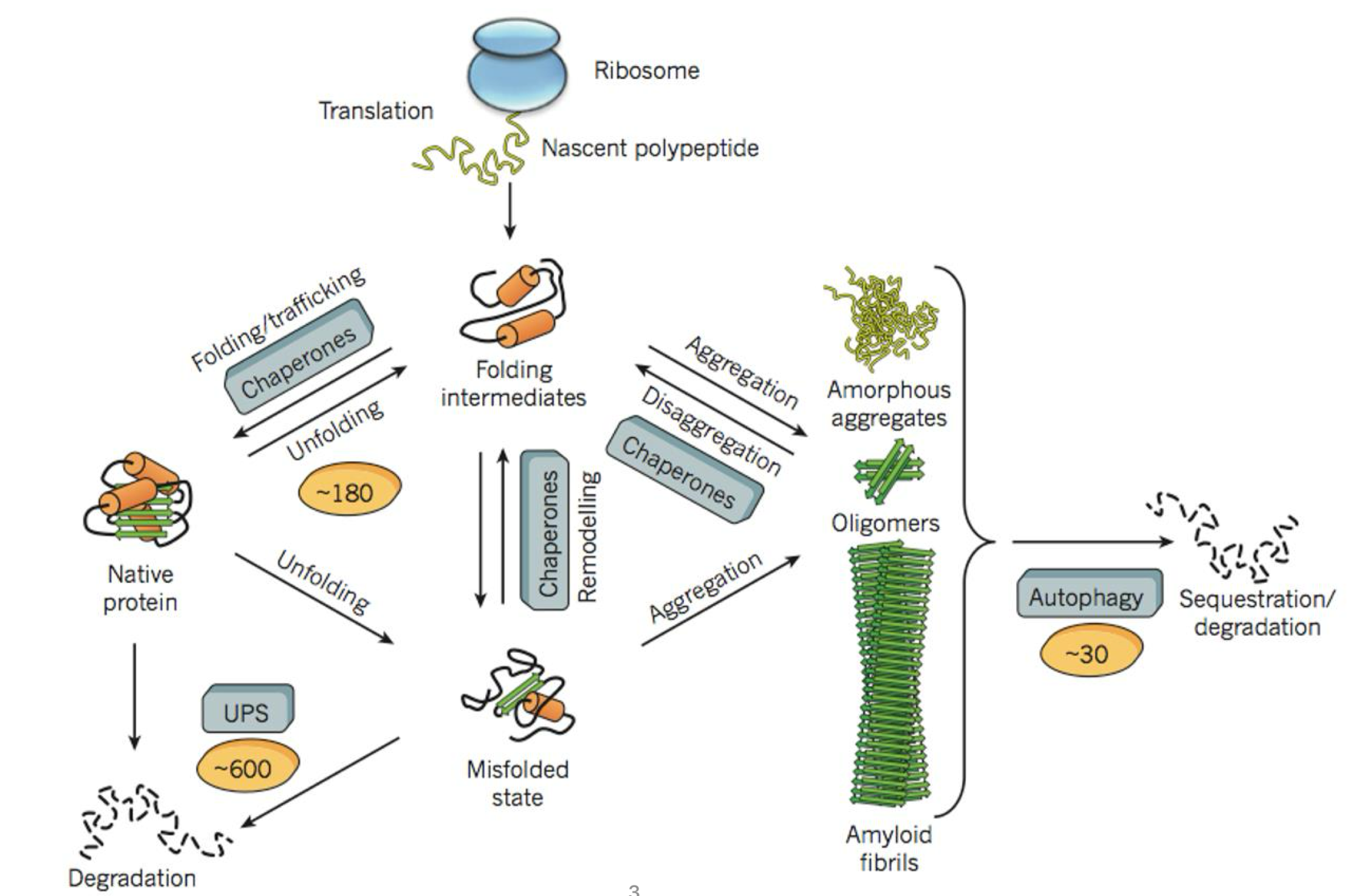
Protein Aggregation & Diseases
there are over 30 human pathologies associated to protein aggregation
e.g. Alzheimer’s (AD), Amyotrophic lateral sclerosis (ALS), Huntington, Parkinson’s, prion diseases
these diseases are characterized by the presence of amyloid protein aggregates in the affected tissues
specific proteins are found at unusually high concentrations inside the aggregates compared to their normal levels.
in most cases, the cause of the disease is unclear and no cure exists
there are many types of amyloid diseases
Amyloid Fibrils vs Amorphous fibrils
amorphous aggregates: disordered protein clumps, usually not associated to disease
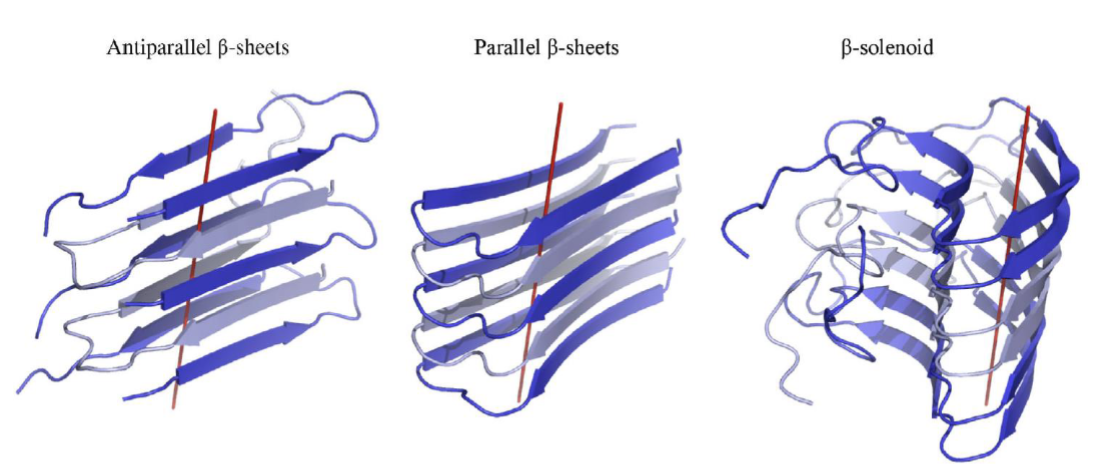
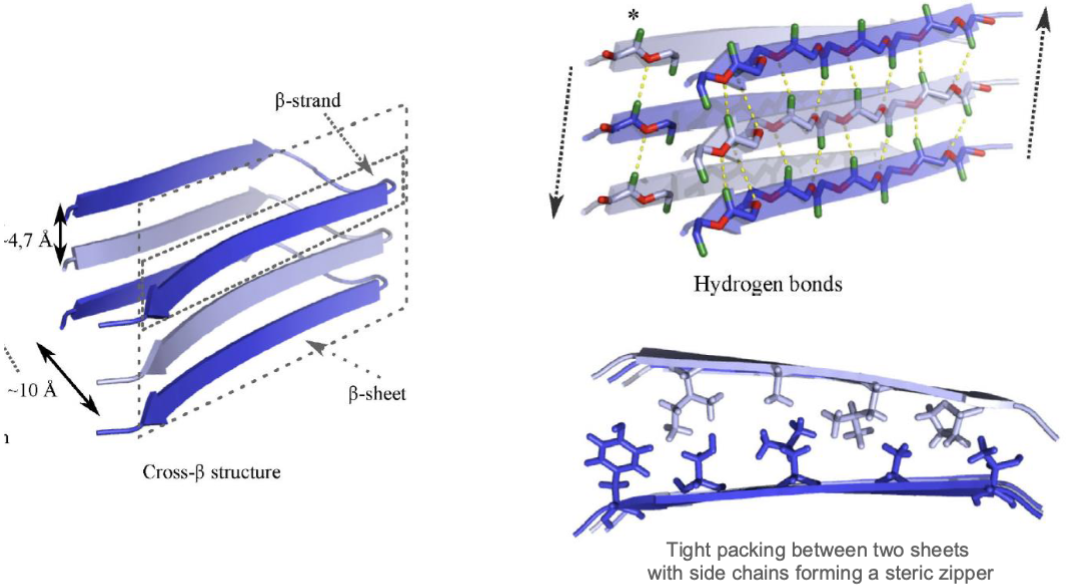
amyloid aggregates: highly ordered fibrils with a characteristic cross-β structure, and are associated to disease, insoluble protein assemblies
β-strands aligned perpendicularly to the fibril axis
cross-β structure refers to the stacked β-sheet arrangement, which provides fibrils w/ high stability, and that an be arranged in 3 manners
antiparallel β-sheets, parallel β-sheets, β-solenoid
high stability => resistant to degradation or resolubilization
Amyloid Fibrils: Amyloid Organization
amyloids are organized hierarchically
starts w/ individual proteins/peptides that form β-sheets (D)
β-sheets stack to form protofilaments (C),
which twist together into amyloid fibrils (B), creating highly stable and structured aggregates (A)
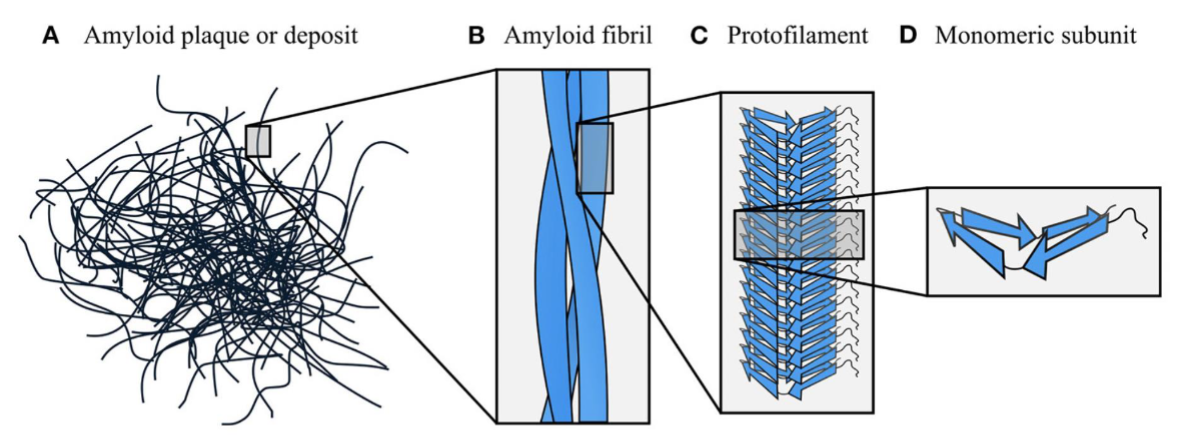
Amyloid Fibrils: Insolubility
amyloid insolubility arises from extensive intermolecular H-bonding between β-strands, which stabilizes the stacked β-sheet structure
and…
hydrophobic interactions, VDW forces and electrostatic interactions further stabilize fibril packing, making amyloids highly resistant to degradation and solubilization
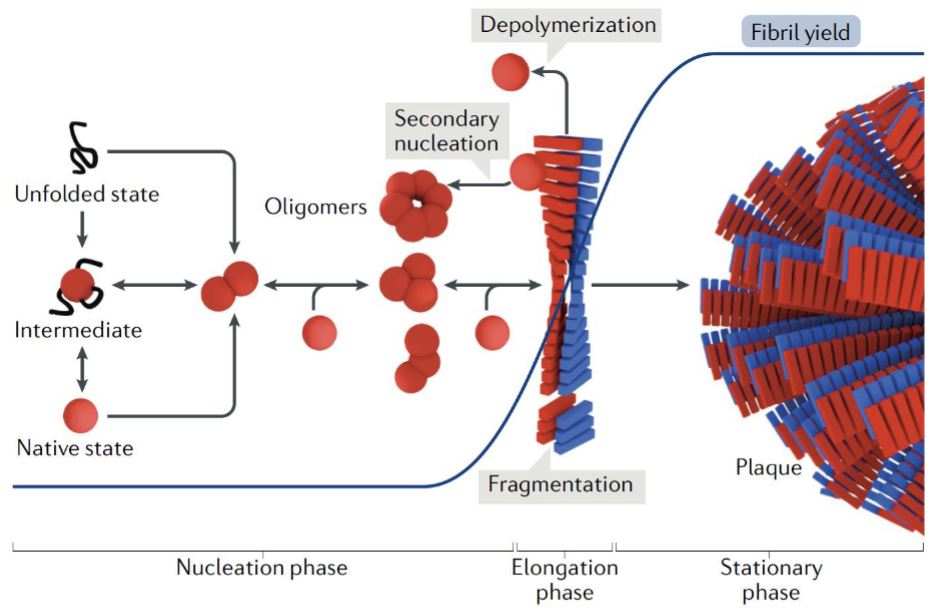
Assembly of Amyloid Fibrils
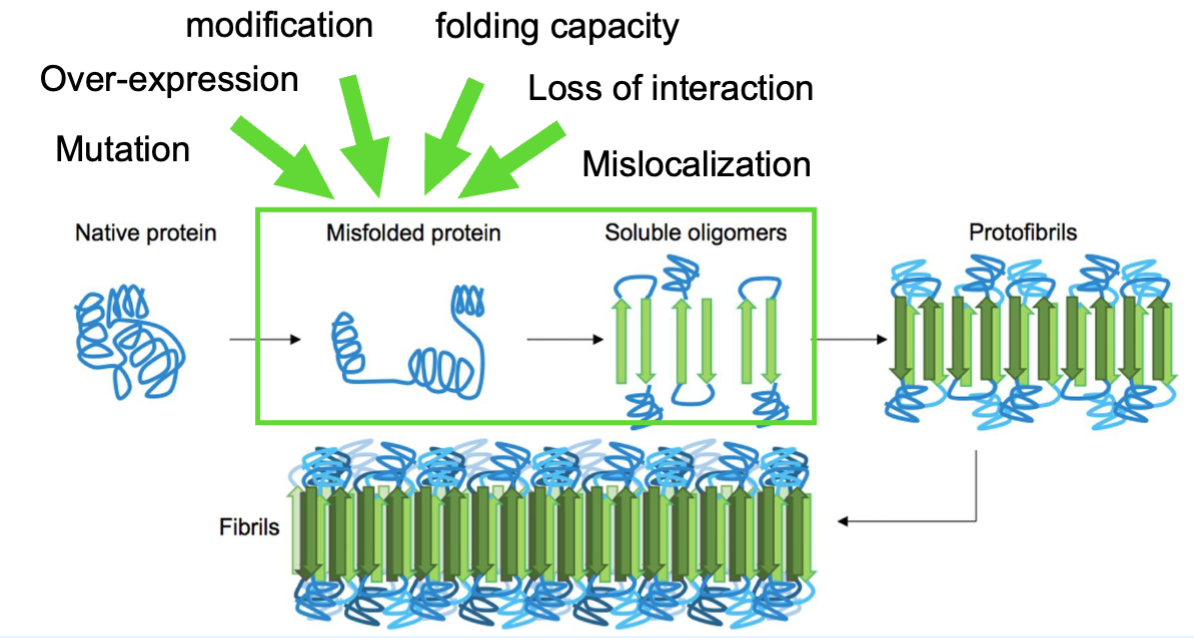
amyloid fibril formation starts w/ misfolded protein or peptide monomers assembling into oligomers (multiple copies of the same proteins)
oligomers act as seeds for further aggregation
once fibrils form, they can accelerate growth by recruiting more monomers and amplifying fibril formation thru fragmentation and/or secondary nucleation, where new fibrils emerge from fibril surfaces
Assembly & Propagation Amyloid Fibrils: Prion
a prion (protein infection) is an amyloid fibril that can convert the native conformation of the PrP prion protein (PrPC) into the infectious nonnative structure (PrPSc) following the formation of a seed
very rare, can be promoted by mutations
only prions are considered infection agents; aggregates formed by other amyloideogenic proteins display prion-like behavior, meaning they can cause template misfolding and spread within and across diff brain regions
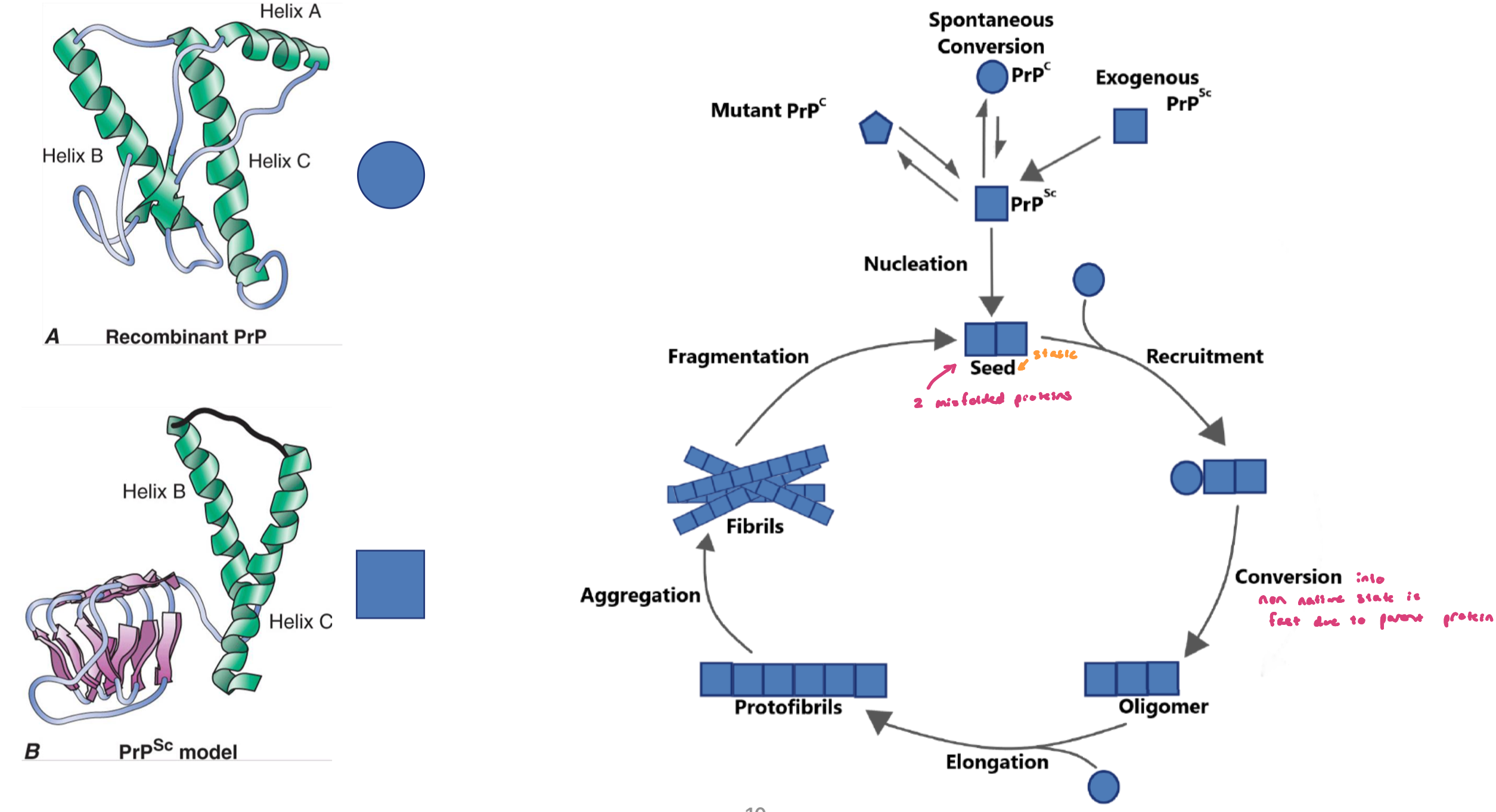
Assembly & Propagation Amyloid Fibrils: Creutzfeldt-Jakob Disease (sCJD)
spontaneous conversion is extremely rare and can cause sCJD disease (infection disease caused by a protein; infection agent = prion protein)
once converted, the nonnative form can propagate thru the brain, leading to rapid dementia
cannabilism and inappropriate handling of surgical tools can transmit the disease, making PrpSc an infection agent that can also infect other mammals
Toxicity of Amyloid Fibrils (diseases)
whether a protein aggregate is toxic or protective has been debated; general agreement is that the small fibrils/protofibrils are cytotoxic
small fibrils: perturbs PM, mitochondrial function (accumulation of reactive oxygen species in cells), physical trafficking by disrupting ER netowrk
can spread from one cell to another (potentially spreading disease over the tissues)
as aggregates get bigger, they start binding and trapping proteins (eg. proteosome)
proteosome normal function: clears misfolded proteins
if trapped by aggregate, it can’t function properly
Amyloid Fibrils: Sporadic Aggregation
sporadic aggregation is rare but many elements can accelerate aggregation (happens spontaneously, w/o a trigger)
Protein aggregation usually requires overcoming an energy barrier (misfolding + nucleation)
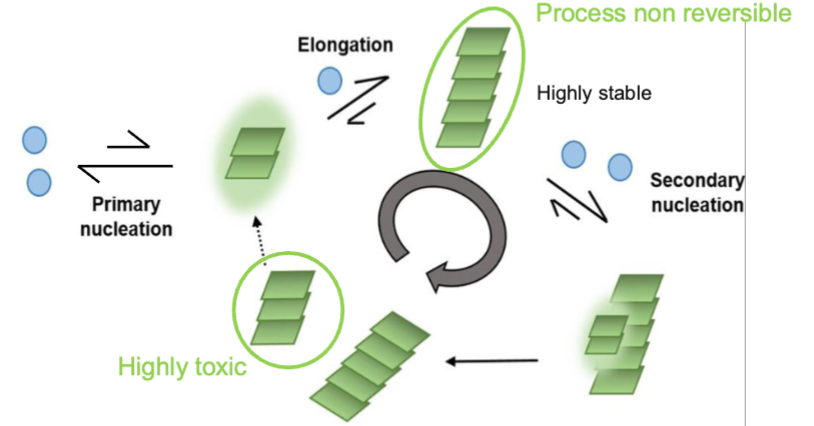
Nucleation
first step and rate-limiting step in amyloid fibril formation
several protein molecules come together to form a small oligemeric nucleus
this nucleus is a rare and short-lived intermediate that must reach a critical size before stable fibril growth can begin
once this nucleus forms, additional proteins can rapidly add to the growing fibril. At this point, the process if non-reversible
in some cases, secondary nucleation can occur; occurs when the surface of existing fibrils promotes the formation of new nuclei, accelerating the production of additional aggregates, including smaller toxic proto-fibrils
Tafamidis
drug to treat transthyretin-mediated amyloid cardiomyopathy
progressive heart condition caused by build-up of transthyretin-containing amyloids in the heart
the small molecule stabilizes the tetramer, reducing the ability of the monomer to misfold and aggregate (need monomer to allow formation of fibrils)