MS & Basis
1/53
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
54 Terms
Définition de la spectrométrie de masse
Mesure le rapport masse/charge (m/z)
Utilise un champ magnétique et/ou électrique pour dévier la trajectoire des molécules chargées et déterminer leur m/z
Architecture d’un spectromètre
4 composants en série
Source : transfert des ions en phase gazeuse
Analyseur : sépare les ions selon m/z
Capacité MS/MS (CID, ETD)
Détecteur : compte les ions et convertit en signaux
Ordinateur : transforme le signal en spectre
Histoire du spectromètre de masse
1943 : Commercialisation du premier spectromètre de masse
1984 : Ionisation Electrospray pour analyser les petites molécules
1885 : MALDI (absorption des composés pour assister l’ionisation laser)
1988 : Ionisation Electrospray de protéines entre 5000 et 40 000 Da + MALDI pour des molécules >10 000 Da
1985 : Commercialisation du premier ESI-MS
1994 : Protéomique
Principes physiques
Loi de Lorentz : F = E . v . B
Un champ E (électrique) et B (magnétique) exerce une force sur une charge individuelle
Loi de Newton : F=m.a
Cominées : (m/Q).a = E . v . B → Le mouvement de l’ion dépend de son rapport m/z
Prix Nobel
Prix Nobel de chimie en 2002 attribué à
Koichi Tanaka : Désorption laser
John Fenn : Electrospray ESI
Pour avoir permis l’ionisation de grosses biomolécules (“making a elephant Fly”
Sources d’ionisation
ESI (Electrospray, Fenn 1988)
Le capillaire amène les molécules de la fin de la chaîne de chromatographie à l’entrée de spectromètre
Une différence de potentiel (champ électrique) est appliqué entre le capillaire et l’entrée → Les molécules sont attirées selon leur polarité
En conditions acides, les molécules chargées positivement sont attirées vers la borne négative (entrée du spectromètre)
Un flux de gaz axial crée des microgoutellettes → vaporisation initiale
L’enceinte chauffée évapore l’eau résiduelle → la densité de charge augmente
La répulsion électrostatique entre charges fragmente les gouttelettes en gouttelettes plus petites (fission de Coulomb)
Résultats : ions en phase gazeuse, chargés par des protons → espèces multichargées
→ Plusieurs protons peuvent se lier de manière non covalente à une même molécule
MALDI (Karas & Tanaka, 1988)
L’échantillon est mélangé avec une matrice organique, puis séché → les molécules de matrice cristallisent et emprissonent les molécules d’intérêt
Un laser IV irradie la matrice (apport d’énergie très rapide) → Les molécules de matrice (cycliques, avec doubles liaisons absorbantes) explosent et transfèrent leur énergie à la molécule d’intérêt
Désorption/ionisation
Résultats : ions monochargés
Collisions
Les collisions avec les molécules de gaz perturbent la trajectoire des ions
→ les spectromètre de masse fonctionnent sous vide pour les éviter
→ L'entrée du spectromètre est très petite ; des pompes maintiennent un vide progressif de plus en plus poussé vers l'analyseur.
Analyseurs
Trois grands types

→ Compromis vitesse/résolution : quadrupôle = rapide mais faible résolution ; Orbitrap = lent mais haute résolution ; TOF = intermédiaire.
Les propriétés et l'usage d'un spectromètre dépendent à la fois de sa source d'ionisation et de son analyseur.
ESI-Q-TOF MS
Instrument hybride combinant source ESI + quadrupôle + TOF pour faire de la MS/MS haute performance.
Architecture (en série) :
Source ESI (Z-spray)
Source ESI orthogonale à l'axe du tube : les neutres vont dans le fond, seuls les ions d'intérêt sont déviés vers l'analyseur.
Hexapôle (ion bridge, transfert)
Quadrupôle (MS1) : filtre de masse, sélectionne l'ion précurseur
Hexapôle (cellule de collision) : fragmentation
TOF (MS2) avec réflectron : analyse les fragments à haute résolution
Quadrupole
4 barres reliées deux par deux avec des polarités alternées.
En modulant la fréquence
On peut laisser passer tous les ions
On peut sélectionner une gamme m/z spécifique. L’ion va osciller à la bonne fréquence et ne se heurtera pas contre la barre. Plus la
Analyse ciblée et quantitative
Cellule de collision
On peut injecter du gaz pour provoquer des collisions
→ Transfert d’énergie vibrationnelle
→ Fragmentation aux liaisons peptidiques
→ Permet de déduire la séquence du peptide
Analyseur TOF (Time of Flight)
Les ions accélérés par un pulse de champ électrique traversent un tube de longueur connue.
Comme la force appliquée et la longueur du tube sont connues, la mesure du temps de traversée donne la vitesse → puis le rapport m/z.
Les ions les plus légers arrivent les plus rapidement
Temps de traversée : 10 000 us
Résolution : 10 000 – 50 000 (avec réflectron).
Capacité : ~100 fragmentations MS/MS par seconde.
Variantes des pièges à ions
Linear ion trap : ions piégés entre des barres du quadrupôle
Les ions résonnants sont éjectés vers le détecteur, les non résonants restent piégés
Quadrupôle ion trap (3D) : piègeage des ions dans un volume confiné par champ quadrupolaire
Les ions sont tous dedans mais seuls les ions instables peuvent sortir
Les 4 barres sont remplacées par deux chapeaux et un anneau
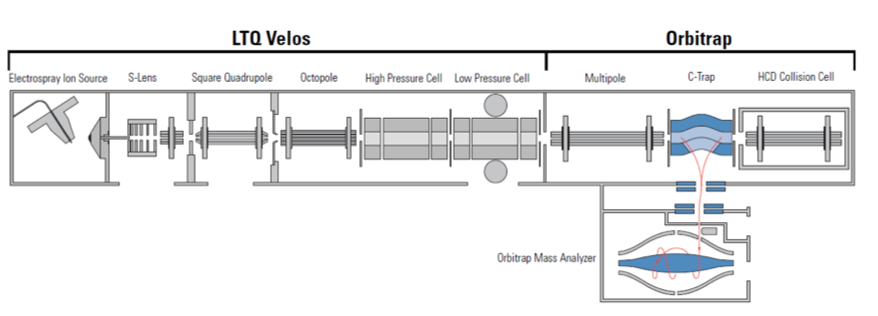
LTQ-Orbitrap FTMS
Instrument hybride couplant un piège à ions linéaire (LTQ Vélos) et un analyseur Orbitrap (détection par transformée de Fourier)
Architecture en série :
LTQ (Velos) :
Source ESI pas dans l’axe
S-Lens (tube pour terminer la déshydratation)
Quadrupôle carré
Octopole pour garder les ions dans un flux
Cellule haute pression (fragmentation)
Cellule basse pression (analyse)
Orbitrap :
Multipole pour garder les ions dans un flux
c-Trap = lentille spéciale qui accumule, accélère et envoie les ions dans l’Orbitrap
Cellules de collision HCD (fragmentation aléatoire)
Orbitrap (détection Ft haute résolution)
Il combine la rapidité/sensibilité du piège linéaire avec l’haute résolution du Orbitrap et permet plusieurs modes de fragmentation (CID dans LTQ, HCD du côté Orbitrap)
Orbitrap
Les ions sont injectés et tournent autour d'une électrode centrale en forme de broche tout en oscillant latéralement.
Piège les ions en mouvement orbital au sein d’un champ électromagnétique
La fréquence d'oscillation latérale est proportionnelle au rapport m/z → résolution très élevée.
Développement de la protéomique
Plusieurs domaines ont convergé vers la protéomique en 1990 :
MS
Electrophorèse 2D
Séquençage des nucléotides/ESTs
Approches génétiques (approches chip-based)
LC-MS(/MS) (analyse de mélanges complexes)
Etapes clés :
1994 : algorithme pour interpréter les spectres MS/MS
1996 : séquençage du génome de la levure
2001 : séquençage du génome humaine
Fragmentation des protéines et des peptides
CID (Collision Induced Dissociation)
Collisions successives avec un gaz inerte
Augmentation progressive de l’énergie interne des ions
Fragmentation des liaisons le plus faibles (squelette peptidique)
Processus dépendant de l'énergie pouvant entraîner la perte de modifications post-traductionnelles labiles (sensibles)
ETD (Electron Transfer Dissociation)
Implique des transferts rapides d’électron à l’ion
Dissociation rapide
Sans redistribution de l’énergie avant fragmentation
Préserve mieux les modifications post-traductionnelles labiles
Caractérisation de protéines pures
La spectrométrie de masse aide à déterminer l’identité, la quantité mais également le repliement (folding) des protéines
Exemple sur le mutant CALR oncogène et son rôle dans la dimérisation du récepteur de la thrombopoïétine
→ Une protéine produite industriellement peut exister sous de nombreuses formes indésirables
PTMs (modifications post-traductionnelles)
Clipping (clivages)
Mutations
Modifications chimiques
→ Cette diversité s’étend sur 5 ordres de grandeurs d’abondance (de 80% à 0,01%) d’où les questions clés
Combien de formes
Leurs abondances relatives ?
Quelles modifications ?
Où sont-elles localisés ?
Pourquoi sont-elles présentes ?
Stratégies pour déterminer l’identité et la quantité des protéines indésirables
Stratégie 2 approches complémentaires :
Bottom-up / Protein sequencing (LC-MS) : digestion enzymatique (ex. trypsine) → analyse des peptides résultants par chromatographie liquide couplée à la MS.
Top-down / Entire protein (MS) : analyse de la protéine intacte (après séparation type SDS-PAGE ou chromatographie) directement par MS.
SDS-PAGE 1D : séparation selon la masse moléculaire apparente.
Gel 2D : 1ère dimension = focalisation isoélectrique (point isoélectrique) ; 2ème dimension = masse → deux paramètres séparateurs.
Utile car certaines formes ont la même masse mais un point isoélectrique différent.
Les peptides sont séparés par chromatographie liquide en phase inverse (C18) puis analysés par spectrométrie de masse.
Analyse des protéines entières et séquençag
Préparation des échantillons pour l’analyse par spectrométrie de masse (MS)
Détermination de la masse moléculaire de la protéine entière
Considérations sur la résolution et le motif isotopique
Exemple : IgG
Traitement des données
Identification des protéines : approche bottom-up
Digestion protéolytique
Identification par séquençage MS/MS
Moteur de recherche Mascot
Méthode optimisée pour l’identification des protéines avec une couverture maximale de séquence
Bases de la MS des biomolécules
Conditions à respecter :
Minimiser les seuls
Bien choisir la matrice
Maintenir une haute pureté
Assurer la solubilité
Avoir assez d’échantillons
Adapter la méthodes aux groupes fonctionnelles
Analyser assez rapidement après synthèse purification
Applicables aux protéines, peptides, glucides, oligonucléotides et petites biomolécules
Préparation des échantillons - purification/échange de tampon
Précipitation : Utilisée pour concentrer les protéines et éliminer les contaminants tels que les sels et les détergents.
Elle est généralement réalisée avec de l’acétone, du TCA ou du méthanol/chloroforme. Le TCA est plus efficace que l’acétone seul pour révéler des spots sur gel 2D.
Le culot protéique est ensuite redissous dans un tampon adapté à la MS.
Ultrafiltration : Utilise des filtres avec différents cut-offs (NMWL) de poids moléculaire spécifiques afin de retenir les protéines tout en laissant passer les petites molécules, en utilisant des tampons volatils compatibles MS (NH₄Ac, NH₄HCO₃).
Méthode efficace pour l’échange de tampon et la concentration des échantillons.
Adsorption (ZipTip) : Repose sur la fixation des protéines à une matrice solide (par ex. billes C18 ou phase inverse C4 selon la taille).
On dépose donc la pointe de la pipette sur un lit de chromatographie
Blind/Wash/Elute en milieu FA ou TFA
Permet la purification, le dessalage et parfois la pré-concentration avant la MS.
Perméation sur gel (chromatographie d’exclusion de taille) : Sépare les protéines selon leur taille moléculaire.
Les grosses molécules sortent en premier et les sels sont retenus → dessalage
Sépare les complexes protéiques
Colonne de centrifugation contenant une matrice de plyacrylamide
Résine d'élimination de détergents : élimine SDS, CHAPS, Triton, etc. avec bonne récupération de la protéine
Les détergents interfèrent avec l’ionisation, notamment en ESI-MS
Séparation par électrophorèse : SDS-PAGE 1D, 2D-PAGE ou HPLC. Bon pour l'identification des protéines, mais inadapté pour la détermination de la masse intacte (dénaturation + digestion en gel).
Résolution d’un pic isolé
R=Δm/m
où :
m = masse de l’ion,
Δm = largeur du pic à mi-hauteur (FWHM, Full Width at Half Maximum).
Plus le R est élevé, plus le pic est fin, mieux les isotopes sont séparés
Exemple de résolution
À basse résolution (ex. 1000), les pics isotypiques se confondent en une enveloppe gaussienne
À haute résolution (>10 000), ils sont individuellement résolus
Quand la résolution augmente :
les pics deviennent plus fins,
ils sont mieux séparés.