L7- Cellular senescence, apoptosis & maintaining genome stability II
1/45
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
46 Terms
what is apoptosis
Allows cell to die in restrictive manner without affecting environment
Contrast to necrosis- accidental cell death
when is apoptosis important
Important in embryogenesis and normal tissue homeostasis
• Provides a self-destruct mechanism for damaged cells and is a crucial anti-cancer mechanism
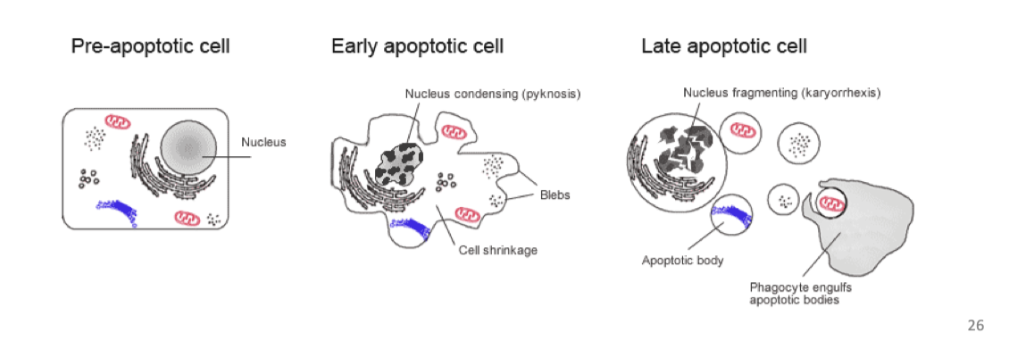
how does apoptosis occur
A series of biochemical events that lead to characteristic cell changes and cell death / “recycling”. These changes include blebbing,
cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation.
Form apoptotic bodies that are then taken up
why does Unrepaired or excessive DNA damage leads to apoptosis
This prevents damaged DNA converting to mutations in progeny cells
i.e. prevents the propagation of deleterious mutations that might otherwise activate oncogenes or inactivate tumour suppressor genes.
why do Inappropriate growth signals lead to appoptosis
Inappropriate growth signals (such as those resulting from oncogene activation) can lead to apoptosis.
So activation of a growth-promoting oncogene can be pro-apoptotic (archetype c-MYC).
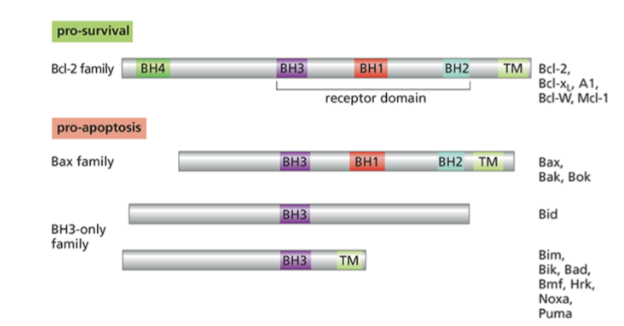
what is the Bcl-2 family
A family of proteins that have BH homology domain that allows them to interact with each other
Cell fate determined by the balance of these family members
Many cancers upregulate antiapoptotic bcl2 proteins
what s the function of BAX (a Bcl-2 protein)
BAX promotes apoptosis by forming oligomers in the outer mitochondrial membrane, creating pores that release cytochrome c and activate caspases.
what are the members of the Bcl-2 family
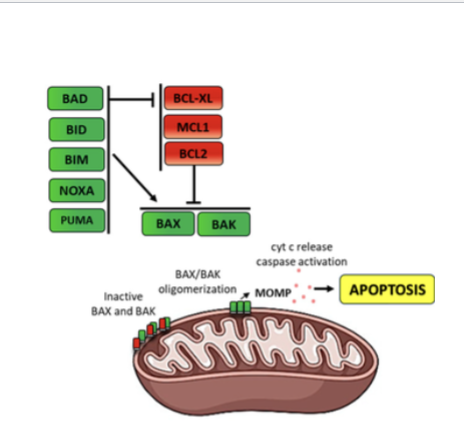
how do Bcl-2 proteins relate to apoptosis
The balance between pro- and anti-apoptotic Bcl-2 family proteins determines whether apoptosis occurs.
Under normal conditions anti-apoptotic proteins inhibit BAX/Bak,
but after DNA damage BH3-only proteins block the anti-apoptotic proteins, allowing BAX/Bak to trigger apoptosis.
what is Venetoclax
a BH3 mimetic
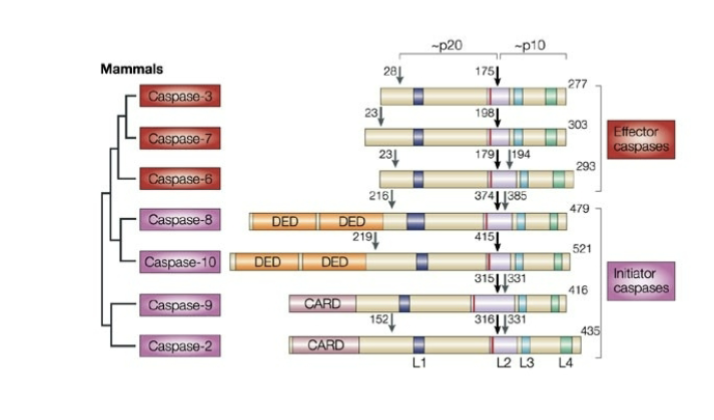
what are caspases
A family of cysteine proteases
Effector caspases require cleavage by initial caspases to become active
initiator caspases- 8, 10, 9, 2
Effector caspases – “executors of apoptosis” 3, 7, 6
what are the 2 key apoptosis pathways
2 key pathways
Intrinsic- important in cancer, intertwined with pathways that sense DNA damage
Extrinsic- linked to extracellular signalling e.g. death signalling. Ultimately lead to activation of intracellular pathway
Cross talk via activation of BID
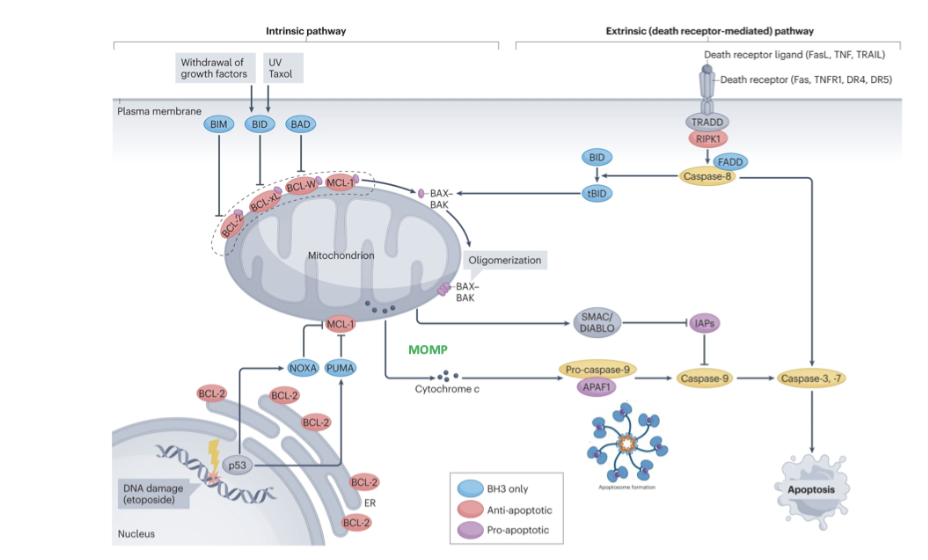
what is the role of SMAC in the apoptosis pathway
inhibits inhibitor of apoptosis proteins- ensures activation of caspase (initiation caspase)
what is the role of the disc complex in the apoptosis pathway
Disc complex- leads to activation of caspase 8 (initiator) that leads to effectors
what is the cell intrinsic apoptosis pathway
DNA damage causes activation of p53
p53 drives expression of BH3 only members (eg puma, noxa
bind and inhibit action of anti apoptotic Bcl-2 proteins leaving BAX and BAX free to form pores/oligomers in outer mitochondrial membrane
cytochrome c released from mitochondria and binds to APAF1
SMAC/diablo also releases, inhibits inhibitor of apoptosis proteins to ensure activation of activator caspase 9
APAf1 binds caspase 9(initiator
activation of initiator caspase to activate effector caspase (3,7)
cleave cell contents, cell shrinks, cell death
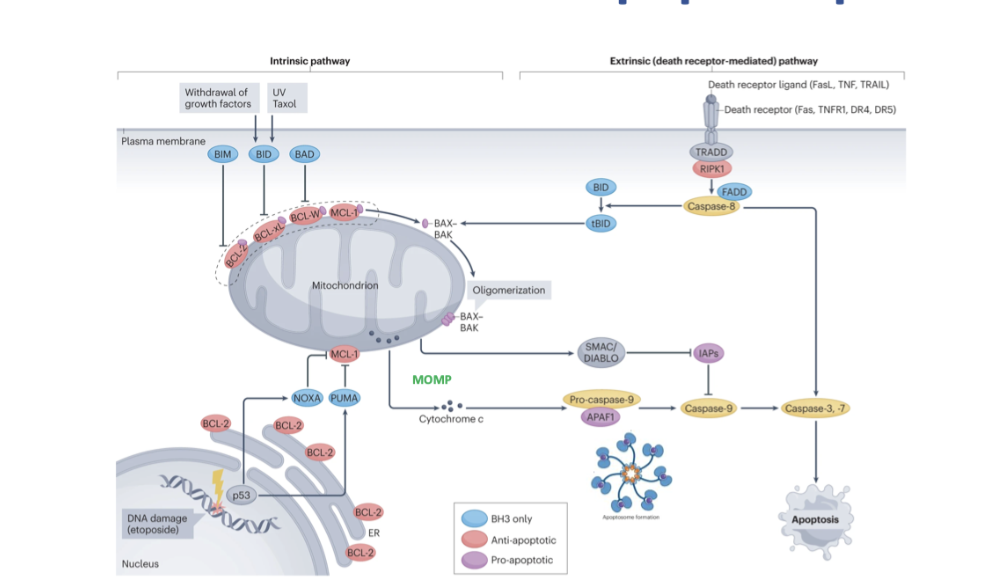
what is the cell extrinsic apoptosis pathway
receptor, ligand and death receptor fuse
forms DISC complex with recruits pro caspases to activate caspase 8
leads to signalling down to effector caspases
cross talk and ability for 2 pathways to join via activation of BID, causing BAX and BAK activation, forming oligomers in outer mitochondrial membrane
what are the most notable triggers of apoptosis
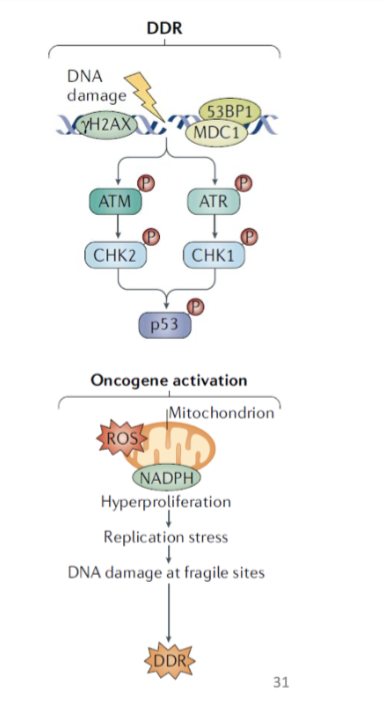
DDR- Cause phosphorylation of p53
hyperactive oncogenic signalling- Due to oxidative stress or ROS
Evading apoptosis is a key step during carcinogenesis and therapy resistance
Numerous mechanisms to resist cell death
what is one main issue of cancer therapies
Fundamental rationale of cancer therapies - DNA damaging
Not selective to only cancer cells
Harms cells that proliferate fast
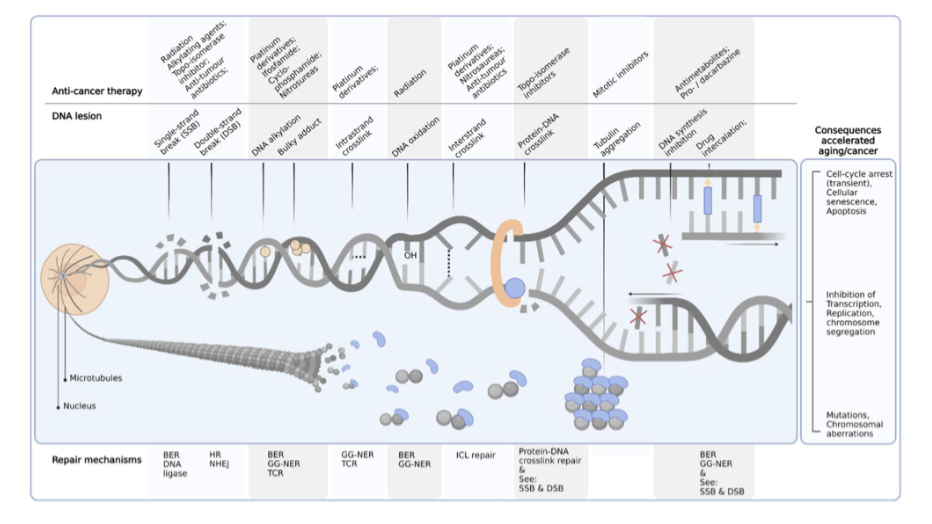
what are some mechanisms to resist cell death
impaired receptor signalling pathway
reduced expression of death receptor and death signal
defects/mutations in p53
disrupted balance of Bcl-2 family
underexpression of proapoptotic proteins
overexpression of antiapoptotic proteins
overexpression of IAP5
reduced caspase activity
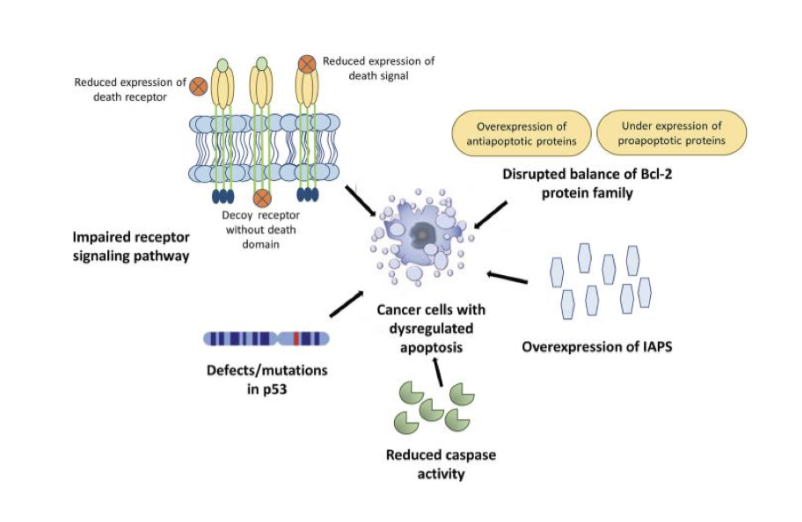
give an overview of the p53 pathway
P53 induces expression of negative regulator mdm2 which drives the degradation of p53- very controlled negative feedback loop
High p14 levels regulates mdm2 levels
Wip1- direct target of p53- phosphatase and removes any phosphates on p53
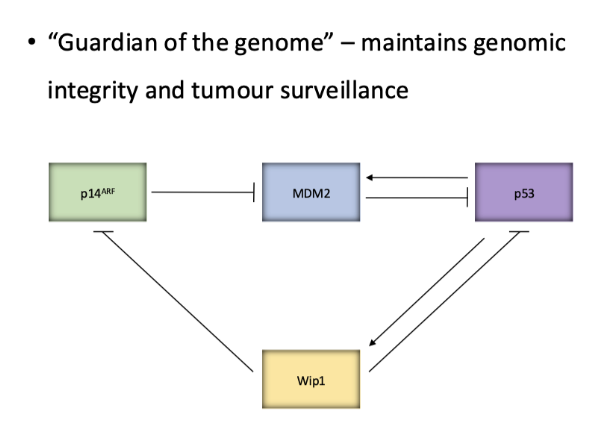
what is the role of ATM and ATR
ATM and ATR signalling to respective proteins CHK1/2 and activate by phosphorylation
Then phosphorylate p53 to prevent it form binding mdm2- activating it
P53 normally has short half life when not activated
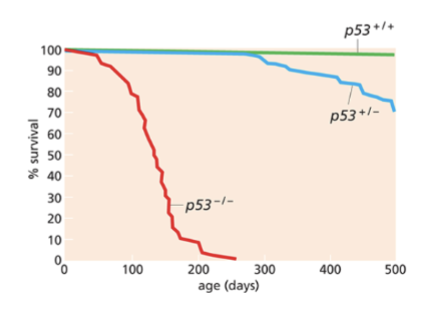
describe the findings from p53 KO mice
p53 KO mice develop tumours ~4.5 months
• mdm2 KO mice die early in development
• Mice deficient for both mdm2 and p53 develop normally and are viable
what are the 4 mechanisms of p53 signalling inhibition
TP53 mutations (~50%)
MDM2/MDMX amplification or overexpression
p14ARF deletion or promoter hypermethylation
WIP1 overexpression
describe the relevance of p53 mutations in cancer
P53 inactivated by mutation in 50% of cancers
These pathways don’t have to happen simultaneously
mutations don’t always inactivate it, may be gain of function that drive cancer development
where is the most common area for p53 mutations
Hotspots normally in DNA binding domain of p53- between exons 5-8
P53 acts as tetramer- has a dominant negative effect
Accumulation of p53 indicates a mutation that increases its half life as mdm2 doesn’t negatively regulate it

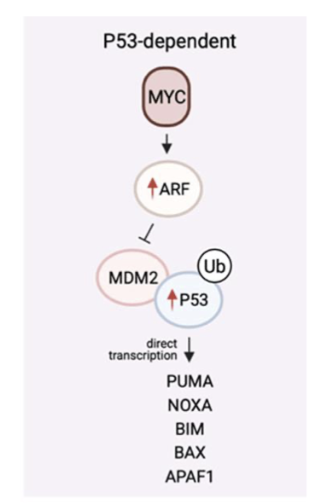
what is the pathway of oncogene induced apoptosis
In response to oncogenic hyperactive signalling- can lead to apoptosis
sent by p14- ARF (aRF upregulation)
upreg of p53
drive apoptosis
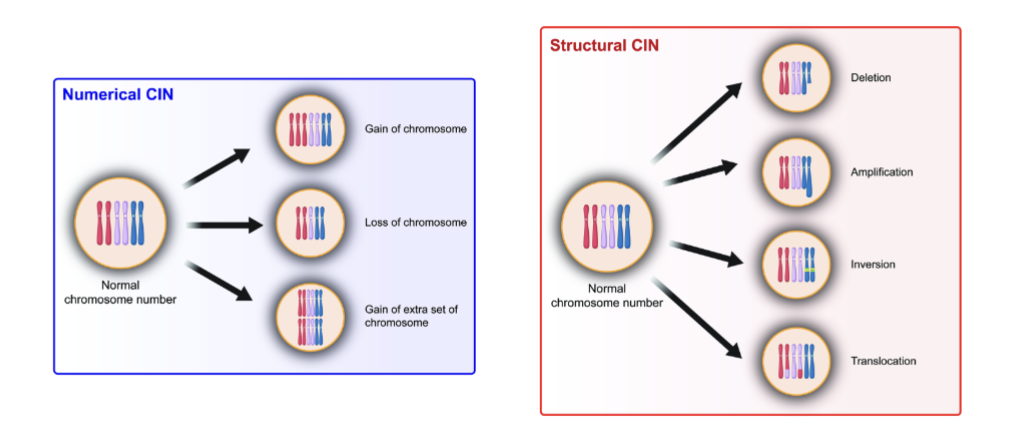
what is genomic instability, what are the 2 forms
Characteristic of most if not all cancers
Accumulation of instability that deviate from the norm
Chromosomal level (CIN) structural and numerical- Gaining a portion or an entire set
Non-CIN (DNA level)
describe non CIN (DNA level) genomic instability
Microsatellite instability (MIN) composed of short nucleotide repeat sequences resulting from impaired DNA mismatch repair
Increased frequency of single/few bp substitutions, insertions, deletions
what are the 2 types of chromosomal instability (CIN)
numerical changes (aneuploidy)
structural changes
vast majority of human tumours exhibit CIN (~60-80%)
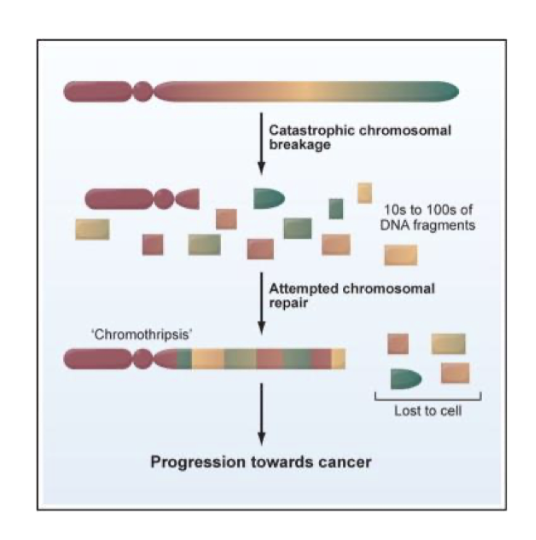
what is chromothripsis
Shattering of one or more chromosomes
All pieces randomly put together
Catastrophic and rare
what are some sources of DNA instability
Sources:
telomere dysfunction
various exogenous and endogenous sources of DNA damage
replication or segregation errors
telomere dysfunction
loss of DDR
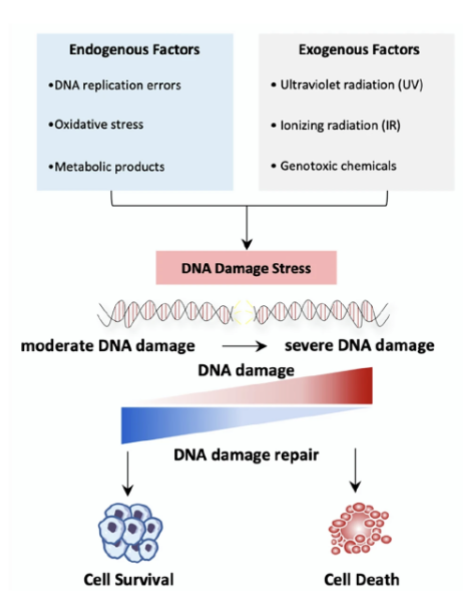
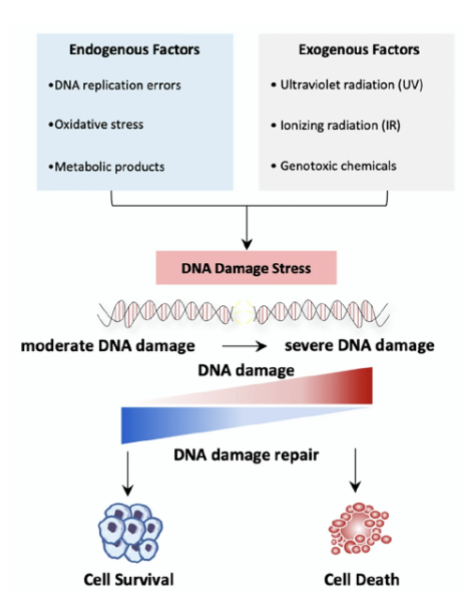
what are the fundamental mechanisms to maintain genomic stability
Activation of DDR
Repair of any DNA damage
Induction of senescence or apoptosis to prevent propagation of cells with damaged DNA
give an overview of DNA repair mechanisms
DSB- most cytotoxic DNA damage- can initiate with translocation and change DNA
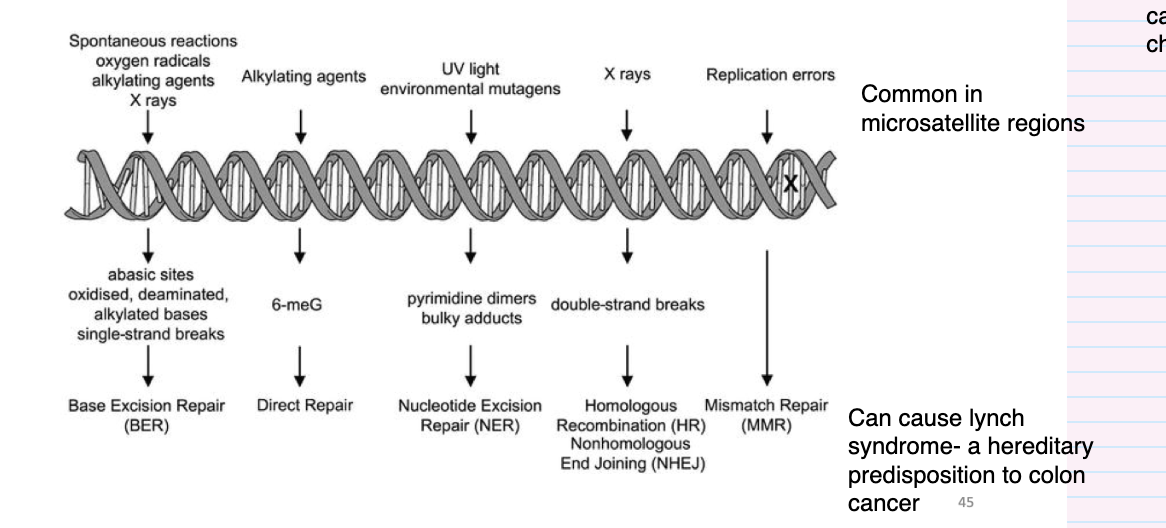
what is mismatch repair (MMR)
Detects any errors in replication
post replicative repair pathway
• Repairs for example base mismatches that have arisen during replication in S phase
• Increased fidelity of replication by 100-fold
• Strand specific
• Aberrations can lead to MIN
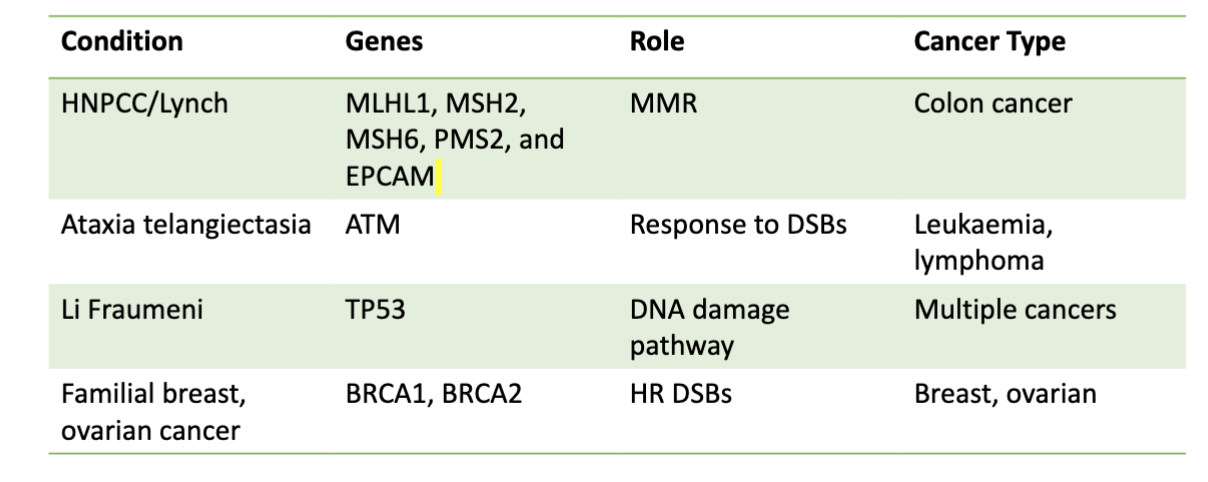
• Lynch syndrome (AKA hereditary nonpolyposis colorectal cancer HNPCC) with increased susceptibility to colon cancer
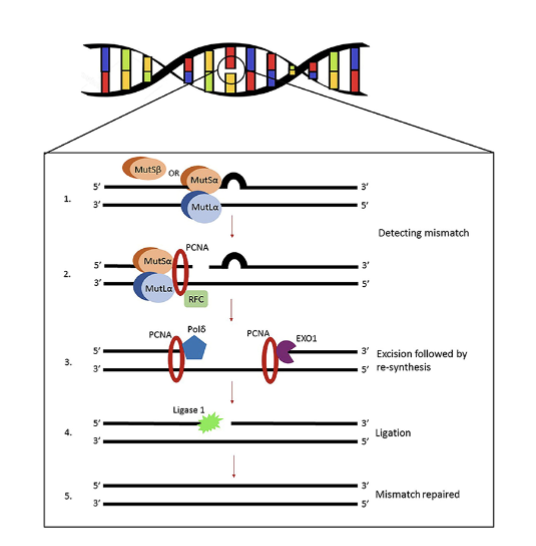
what is the process of MMR
MutSⱭ detects change and recruits MutLⱭ
recruits additional proteins to cleave mismatch
elongation with DNA polymerase
then ligated
Must be able to recognise newly synthesised (inaccurate) strand
why are MutSⱭ and MutLⱭ given these names
called mut due to homology with E.coli proteins where originally studied
what are MutSⱭ and MutLⱭ made up of
MutSⱭ- heterodimers of Msh2,3 and 6 proteins
MutLⱭ- heterodimers of mlh1 and pms 2
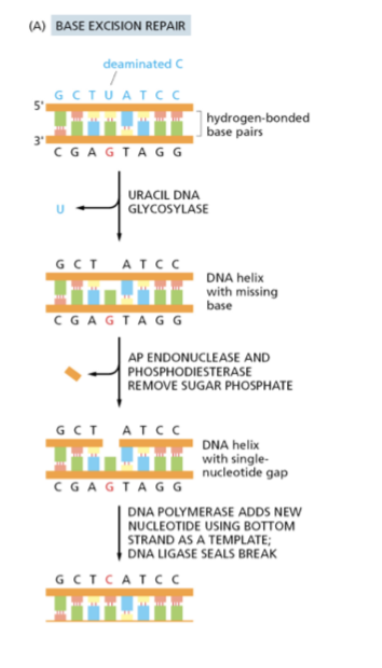
What is base excision repair (BER)
BER remove inaccuracies to the bases themselves- has glycosylases specific to the bases
Generates a ssDNA break- so the single strand base repair mechanism works in tandem
What is nucleotide excision repair (NER)
Much more broad spectrum of damage
repair bulky lesions which distort the DNA- defective in Xeroderma
Pigmentosum
Pyrimidine dimer that causes distortion
Recognised and cleaved
DNA polymerase recruited to synthesis new section, then ligated
On new synthesised strand
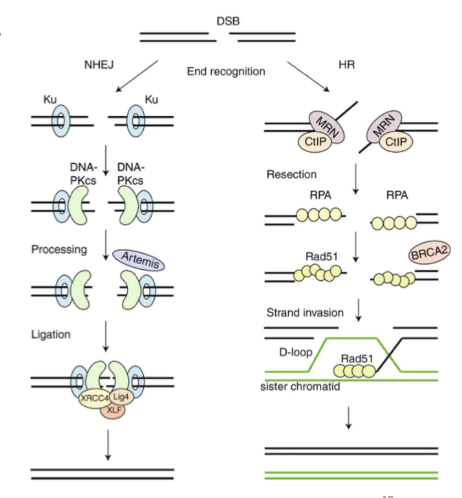
what are the 2 types of DSB repair
NHEJ and HR
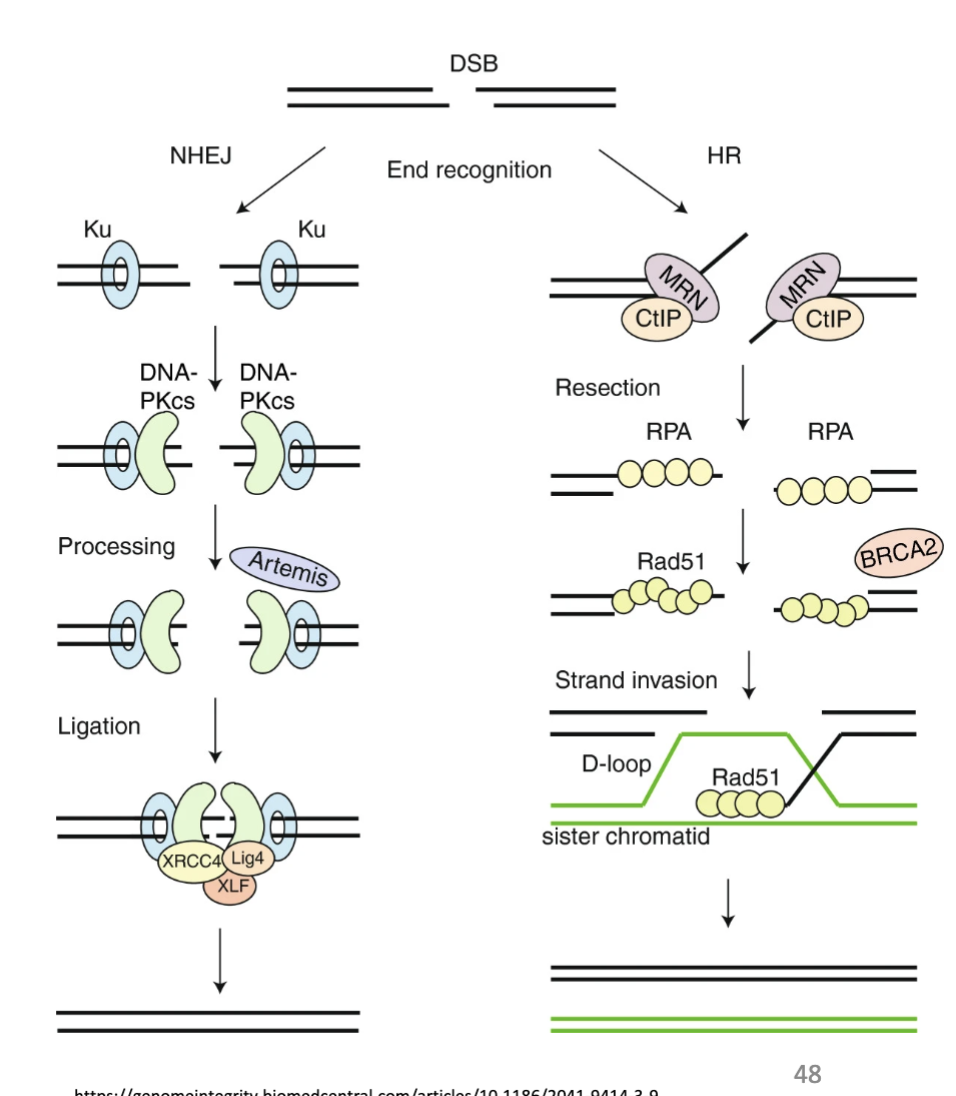
what are some features of DBS repair
DSBs most cytotoxic lesions
Caused by cancer therapies and stalled and collapsed replication forks
2 main pathways: Homologous recombination (HR) and Non-homologous end-joining (NHEJ).
Vary in fidelity of repair and phase of cell cycle
BRCA1/2 and HR: hereditary risk of breast and ovarian cancer
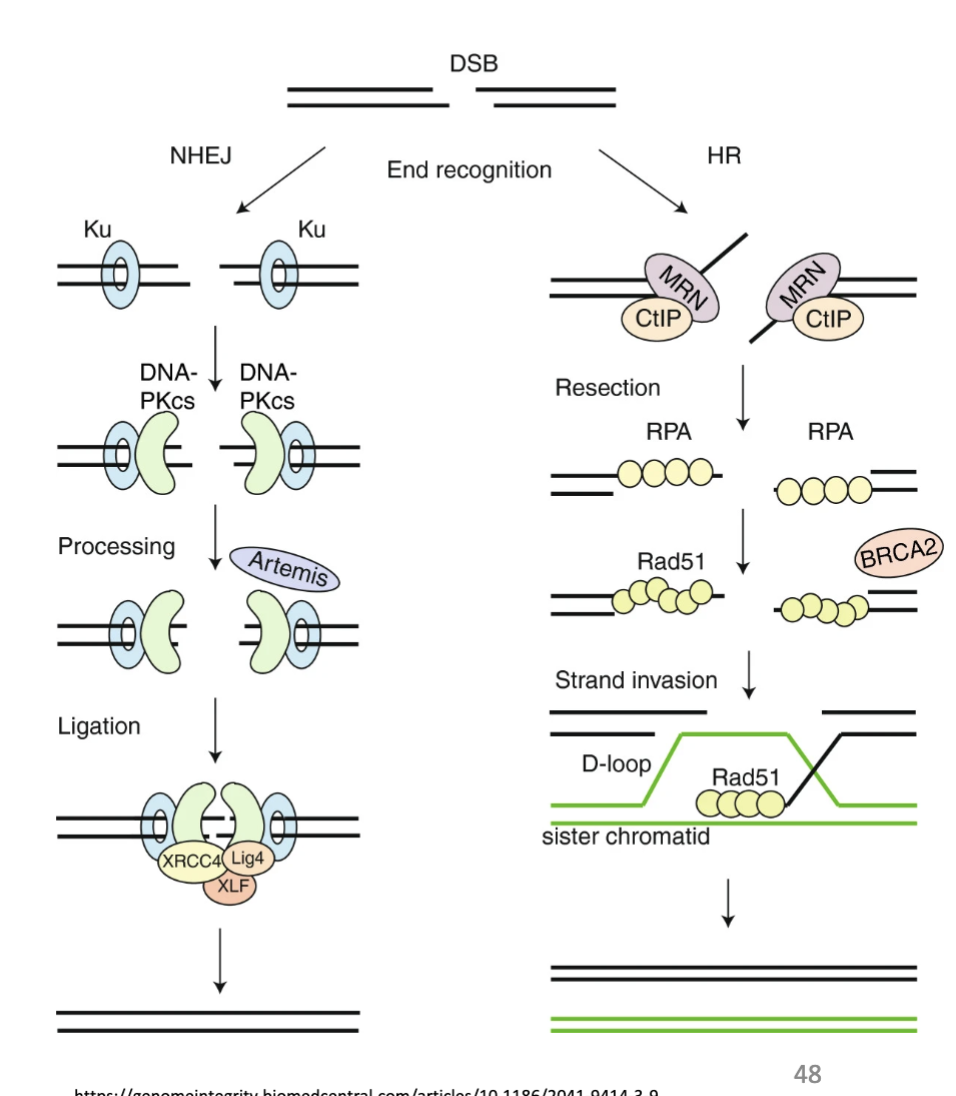
what does DBS occur in response to
Can occur in response to ionisation and stalled replication forks
Fork can be fragile, if held for a long time can break
A ss break can cause a ds break, which can then cause a translocation
describe NHEJ DSB repaur
NHEJ- large proteins detect DSB, processing to remove any single strands
2 ends are simply ligated together
Can happen anywhere in cell cycle
Not as high fidelity- joined whether they should or not
That’s why end of telomeres are protected
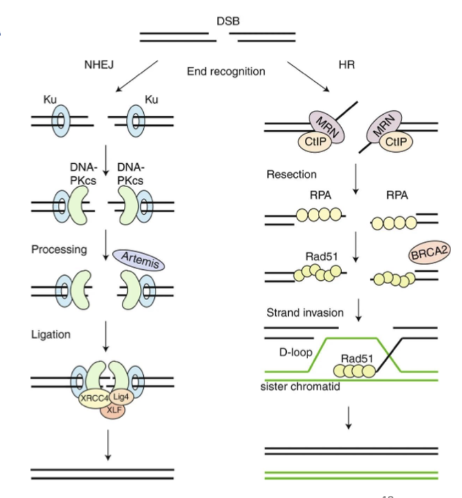
describe HR DSB repair
HR in S or G2 phase- use info from sister chromatid
Recruitment of BRCA and Rad51- causes invasion of sister chromatid so an be used as a template
Higher fidelity
what are some examples of hereditary cancers
Mutations in caretaker genes leads to increased cancer risk due to greater risk of DNA mutations
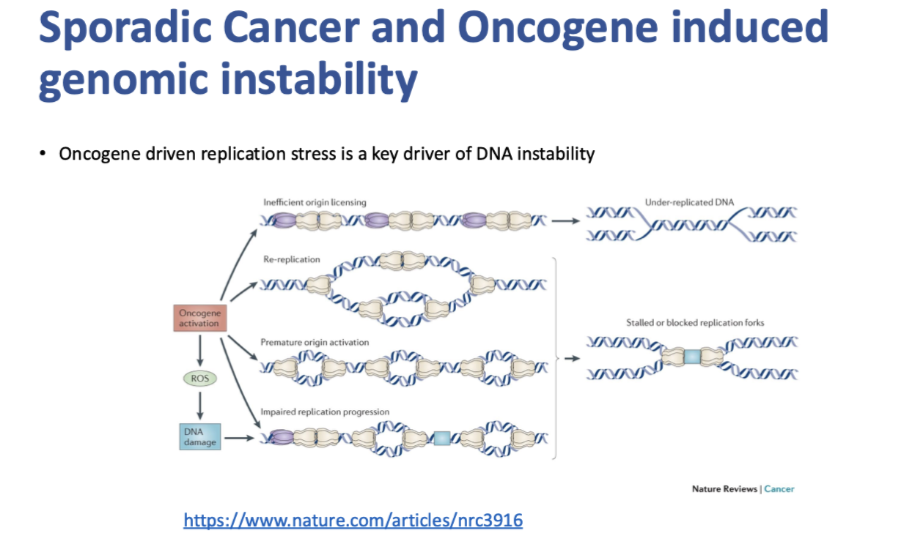
how is oncogene driven replication stress a key driver of DNA instability
Key initiator of genomic instability in sporadic cancer is oncogenic driven replication stress
Stalling replication forks
Replication forks crashing with transcription mechanisms