EXAM 3 Study Guide CHAT
1/116
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
117 Terms
Describe the regulatory steps of the GAL1 Promoter activation and repression system
Repressed state (no galactose): 1.) Gal4 is bound to the UAS_GAL. 2.) Gal80 binds Gal4 and blocks its activation function. 3.) Even though Gal4 is on the DNA, transcription stays off or very low because the transcription machinery is not efficiently recruited.
Activated state (galactose present, glucose absent): 1.) Gal3 senses galactose. 2.) Activated Gal3 interacts with Gal80, preventing Gal80 from inhibiting Gal4. 3.) Gal4 is now free to activate transcription. 4.) Gal4 recruits coactivators, Mediator/chromatin-remodeling activities, general transcription factors, and RNA polymerase II to the core promoter/TATA box. 5.) GAL1 transcription is turned on strongly.
Glucose repression (glucose present): 1.) Mig1 becomes active in the presence of glucose. 2.) Mig1 binds glucose-responsive repression regions in the GAL1 promoter. 3.) Mig1 recruits the Ssn6–Tup1 corepressor complex. 4.) This prevents efficient transcription, so GAL1 is repressed, even if galactose is around.
Describe the cis elements involved in the regulatory steps of the GAL1 Promoter activation and repression system
There are four important cis elements involved in the regulatory steps of the GAL1 Promoter activation system: UAS_GAL (upstream activating sequence), Core promoter / TATA box, Transcription start site (TSS), and Glucose-repression elements. The UAS_GAL (upstream activating sequence) contains Gal4-binding sites, is the main positive regulatory element, and activates transcription. The Core promoter / TATA box is where the general transcription machinery assembles. The Transcription start site (TSS) is where RNA synthesis/transcription begins. Glucose-repression elements are sites that help mediate repression when glucose is present, mainly through repressors such as Mig1.
Describe the trans-acting factors involved in the regulatory steps of the GAL1 Promoter activation system
There are six important trans-acting factors involved in the regulatory steps of the GAL1 Promoter activation system: Gal4, Gal80, Gal3, General transcription factors + RNA polymerase II:, Mig1, Ssn6–Tup1. Gal4 is the sequence-specific activator that binds the UAS_GAL. Gal80 is the repressor that binds Gal4 and blocks its activation domain. Gal3 is the galactose sensor/transducer that relieves Gal80 inhibition in the presence of galactose. General transcription factors + RNA polymerase II assemble at the core promoter to start transcription. Mig1 is a glucose-dependent repressor. Ssn6–Tup1 is the corepressor complex recruited by Mig1.
Explain the role of activators, how they influence transcription as well as the chromatin structure.
Activators are regulatory proteins that bind specific DNA control elements such as enhancers or upstream activating sequences. Their main role is to increase transcription of a target gene. Activators influence transcription by help recruit or stabilize: mediator, general transcription factors (ex. TFIID), RNA polymerase II. This makes assembly of the preinitiation complex (PIC) easier and increases transcription initiation. Activators can influence chromatin structure by often recruiting co-activators that modify chromatin. These can include histone acetyltransferases (HATs), which acetylate histones, and chromatin-remodeling complexes, which reposition or loosen nucleosomes. This makes chromatin more open and accessible, so the transcription machinery can reach the DNA more easily.
Explain the role of co-activators, how they influence transcription as well as the chromatin structure.
Co-activators are proteins that help activators increase transcription, but they usually do not bind DNA directly. They are recruited to genes by DNA-bound activators. Co-activators influence transcription by helping connect activators to mediator, general transcription factors, and RNA polymerase II. This helps assemble or stabilize the preinitiation complex (PIC), increasing transcription. Co-activators influence chromatin structure by often having/recruiting histone acetyltransferase (HAT) activity and chromatin-remodeling complexes. These activities loosen chromatin by acetylating histones or repositioning nucleosomes. This makes DNA more accessible to the transcription machinery.
Explain the role of repressors, how they influence transcription as well as the chromatin structure.
Repressors are regulatory proteins that decrease or shut down transcription of a gene. They usually bind specific DNA regulatory elements such as silencers or repressive control regions. Repressors influence transcription by blocking activators, preventing the recruitment of Mediator, general transcription factors, or RNA polymerase II, and interfere with formation of the preinitiation complex. This lowers or prevents transcription initiation. Repressors influence chromatin structure by often recruiting co-repressors that bring histone deacetylases (HDACs) chromatin-remodeling/compacting complexes. These make chromatin more condensed and less accessible. Tighter chromatin makes it harder for transcription machinery to reach the DNA.
Explain the role of co-repressors, how they influence transcription as well as the chromatin structure.
Co-repressors are proteins that help repressors shut down transcription, but they usually do not bind DNA directly. They are recruited to DNA by DNA-bound repressors. Co-repressors reduce transcription by blocking recruitment or activity of Mediator, general transcription factors, and RNA polymerase II. They also reduce transcription by helping prevent formation of the preinitiation complex. Co-repressors influence chromatin structure by often recruiting or containing histone deacetylases (HDACs) and chromatin-remodeling/compacting complexes. These remove activating histone acetylation and promote tighter nucleosome packing. This makes chromatin more closed and less accessible to transcription machinery.
Explain the role of the Plus 1 site (+1) in transcription
The +1 site is the transcription start site (TSS) and it is the first DNA base that is copied into RNA by RNA polymerase. It marks the point where transcription begins and it determines the 5′ end of the RNA transcript. It matters because it defines exactly where RNA polymerase starts RNA synthesis and its position helps determine the length/sequence of the 5′ untranslated region (5′ UTR) and the rest of the transcript.
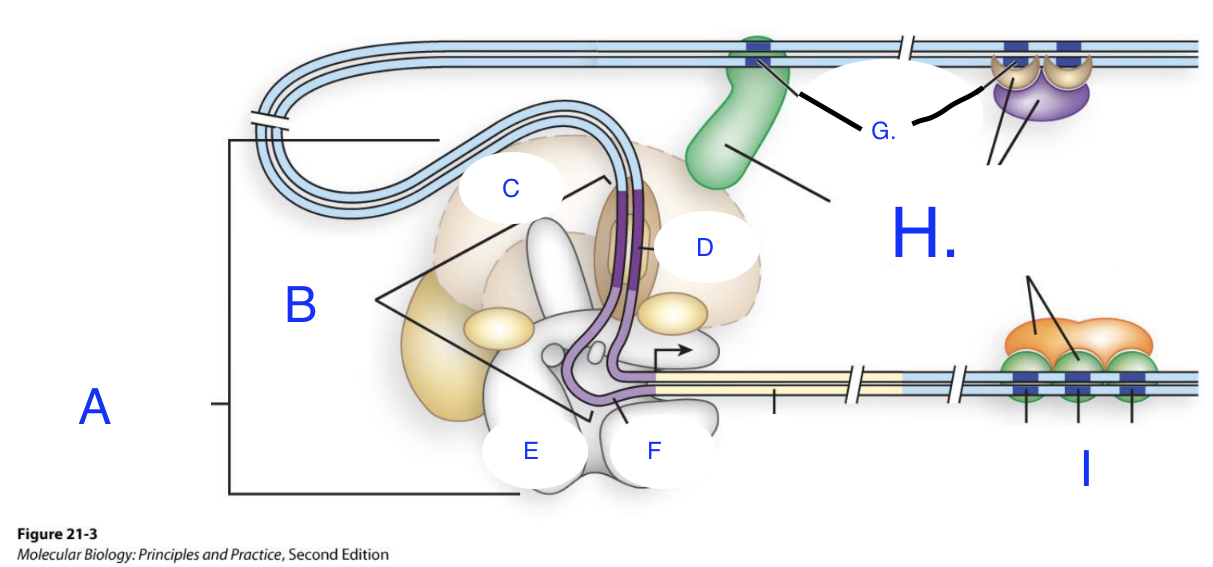
Match the letters in the diagram to parts of gene for transcription below (hint: some of the terms below may or may not be repeated) :
Regulatory Sequences
Transcription activators and co-activators
Gene
Promoter
TATA
POl II
General transcription factors
Inr (the initiator element)
General Transcription Factors
TFIID
A: General transcription factors
B: Promoter
C: TFIID
D: TATA
E: PolI II
F: Inr (the initiator element)
G: Regulatory Sequences
H: Transcription activators and co-activators
I: Regulatory Sequences
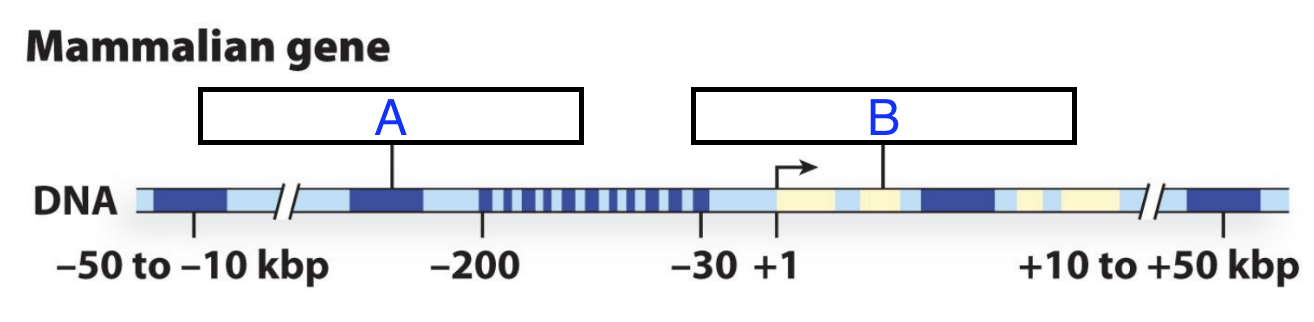
What is the part of the gene for transcription of A and B?
A: Regulatory Region
B: Coding Sequence
Define the regulatory region of a gene and explain how to be able to point out where it is on the gene (diagram)
.
The regulatory region of a gene is the region of a gene where RNA Polymerase and other accessory transcription modulator proteins bind and interact to control RNA synthesis. It usually includes the promoter. Think of a gene like this: upstream DNA -> regulatory region -> +1 start site -> transcribed region -> coding region -> downstream DNA. To identify it on a diagram, if the +1 site, the regulatory region is usually to the left/upstream. The following are clues that you are looking at the regulatory region: TATA box, CAAT box, GC box, UAS, enhancer, silencer.
Define the promoter of a gene and explain how to be able to point out where it is on the gene (diagram)
The promoter of a gene is the DNA region where the transcription machinery assembles to begin transcription. It provides the binding site for RNA polymerase and general transcription factors. It controls whether the gene is transcribed weakly, strongly, or not at all. The main idea for finding the promoter on a gene is that if you can find +1 site, the promoter is usually immediately before it.
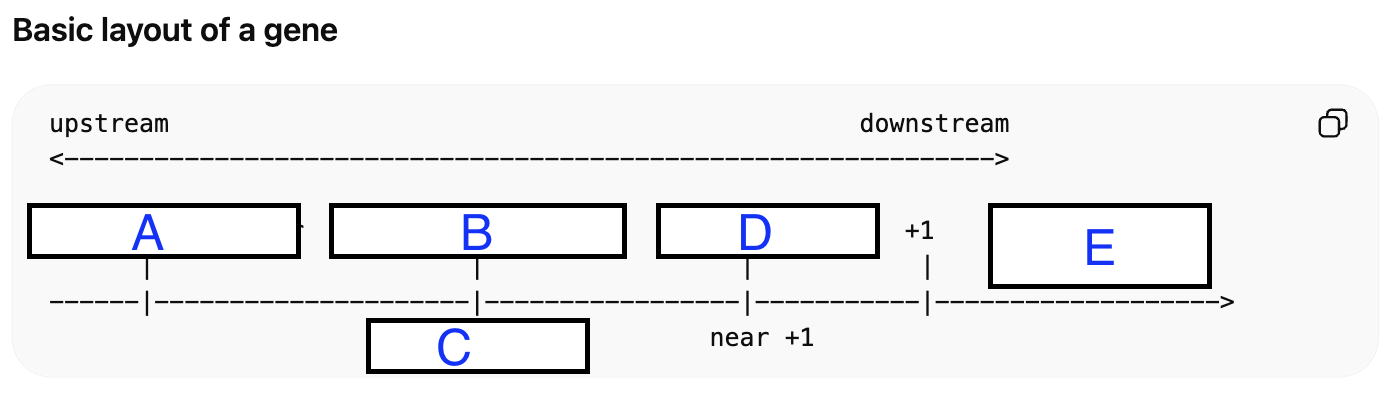
Match the letter with the following regulatory regions below (hint: some terms may be repeated):
Proximal --> Core
Terminator
Enhancer/Silencer
Promoter
Regulatory Sequence
A: Enhancer/Silencer
B: Regulatory Sequence
C: Promoter
D: Proximal / Core
E: Regulatory Sequence
F: Enhancer/Silencer
G: Terminator
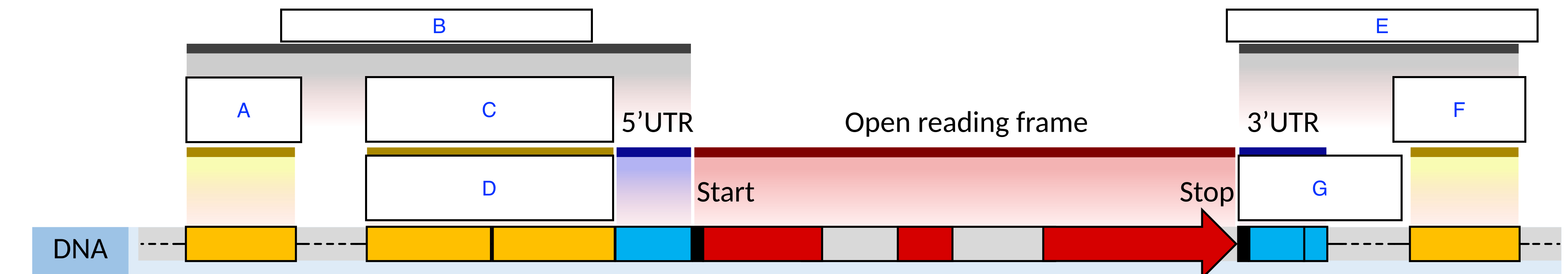
Match the letters with the following regulatory regions/elements bellow (hint: some terms may be repeated):
TATA BOX
Enhancer/Silencer
Proximal Promoter
Core Promoter
Gene body exons / introns
A: Enhancer/Silencer
B: Proximal Promoter
C: TATA BOX
D: Core Promoter
E: Gene body exons / introns2
Define the following regulatory region on the gene and its role: core promoter
The core promoter is the DNA region immediately around the transcription start site (+1). This is where RNA polymerase and general transcription factors assemble. It often contains elements like the TATA box, Inr, or other core promoter motifs. Its role is starting transcription.
Define the following regulatory region on the gene and its role: proximal promoter
The proximal promoter is a short region upstream of the core promoter and it contains binding sites for specific transcription factors. Its role is to increase transcription by serving as binding sites for transcription factors (e.g., CAT box, GC box) that load and stabilize RNA polymerase II. Conversely, their role is also to decrease transcription when repressor proteins bind to them, obstructing RNA polymerase or causing promoter-proximal pausing and premature termination.
Define the following regulatory region on the gene and its role: enhancers
Enhancers are regulatory DNA sequences that bind activators.They can be located upstream, downstream, or inside introns. They can work even when they are far from the promoter. Enhancers’ role is to increase transcription by acting as binding sites for activator proteins, which fold DNA to bring these activators into contact with the promoter, facilitating the recruitment and stabilization of RNA polymerase II. These distant DNA sequences act in three-dimensional space, utilizing mediator complexes to create chromatin loops, bringing the enhancer and gene closer to boost transcription initiation frequency.
Define the following regulatory region on the gene and its role: silencers
Silencers are regulatory DNA sequences that bind repressors. They can be located upstream, downstream, or inside introns. They can work even when they are far from the promoter. Silencers’ role is to Silencers decrease transcription by binding repressor proteins, which physically block RNA polymerase from binding to the promoter, compete with activator proteins, or promote condensed chromatin (heterochromatin) structure, making DNA inaccessible.
Define the transcription initiation site of a gene and explain how to point it out on a gene
The transcription initiation site of a gene, also called the transcription start site (TSS) or +1 site, is the exact DNA nucleotide where RNA polymerase begins making the RNA transcript. The main idea behind being able to point it out on a gene, is that it is just downstream of the promoter, just after the core promoter elements, but before the rest of the transcribed region.
Define the transcription termination site of a gene and explain how to point it out on a gene
The transcription terminator site is the DNA sequence or region that signals RNA polymerase to stop transcription and end the RNA transcript. The main idea behind pointing it out on a gene, is to find the +1 site where transcription starts, follow the gene in the direction of transcription, and the terminator is at the far downstream end, where transcription stops.
How to identify a consensus sequence given a sequence alignment and provide the percent conservation.
For each column: 1.) Count how many A, T, G, C there are. 2.) Pick the most common one -> that is the consensus residue. 3.) Percent conservation =(number of sequences with the most common residue at that position / total number of sequences compared at that position) * 100
What doe the following notions in writing consensus sequences mean?
Y, R, N, ~/~
Y: Pyrimidines = equal T and C
R: Purines = equal A and G
N: no particular base is more common
C/G or C/A or T/A or T/G: one pyrimidine and one purine are equally common
Define the role and characteristics of the -10 region of the prokaryotic promoter during transcription
The -10 region of a prokaryotic promoter, also called the Pribnow box, is a cis-acting DNA element located about 10 base pairs upstream of the transcription start site (+1). It’s role is being a key site recognized by the sigma factor of RNA polymerase. Its main function is to help the DNA unwind (melt) so transcription can begin. This happens because the -10 region is usually A/T-rich, and A-T base pairs separate more easily than G-C base pairs. Its most important characteristic is that it has the following consensus sequence: TATAAT.
Define the role and characteristics of the -35 element of the prokaryotic promoter during transcription
The -35 element is a cis-acting DNA sequence in a prokaryotic promoter located about 35 base pairs upstream of the transcription start site (+1). Its role is serving as an important recognition and binding site for the sigma factor of RNA polymerase. Its main job is to help RNA polymerase initially recognize the promoter and bind in the correct orientation. Its most important characteristic is that it has the following consensus sequence: TTGACA
What are the two forms that prokaryotic RNA polymerase exists as?
The core enzyme and the holoenzyme
Define the holoenzyme form of prokaryotic RNA polymerase and its function
The holoenzyme form of prokaryotic RNA polymerase is the complete promoter-recognizing form of the enzyme. It consists of the core enzyme and a sigma factor. The holoenzyme’s main function is to recognize specific promoter sequences and initiate transcription at the correct site. The sigma factor allows the enzyme to bind promoter elements such as the -35 and -10 regions. Once transcription begins, the sigma factor often dissociates or becomes less tightly associated.
Define the core enzyme form of prokaryotic RNA polymerase and its function
The prokaryotic RNA polymerase core enzyme is a multi-subunit complex responsible for the catalysis of RNA synthesis during transcription elongation. Lacking the sigma subunit, the core enzyme cannot initiate transcription at promoters but efficiently elongates RNA chains by moving along DNA and synthesizing RNA from a DNA template. The function of the The core enzyme is to carry out the catalytic synthesis of RNA from a DNA template. It can unwind and track along DNA during transcription, as well as join ribonucleotides together to make the RNA strand.
What are the components that make up the core enzyme form of prokaryotic RNA polymerase and their respective functions?
The core enzyme is made up of 2 alpha subunits, 1 beta subunit, 1 beta prime subunit, 1 omega subunit. The alpha subunits are essential for the assembly of the enzyme, and the two alpha subunits act as a scaffold. The C-terminal domains of the alpha subunits interact with various regulatory transcription factors to enhance or inhibit gene expression. The beta subunit contains the catalytic site for RNA synthesis and is responsible for binding the incoming ribonucleotide triphosphates (NTPs) and forming the phosphodiester bonds. The beta prime subunit is responsible for DNA binding, and acts like a "clamp" that holds the DNA template securely in the channel so the beta subunit can transcribe it. The role of the omega subunit is not strictly necessary for catalysis, but it is vital for chaperoning. It helps the beta subunit fold correctly and keeps the entire complex stable.
Define sigma factors as they relate to prokaryotic RNA polymerase and its role
Sigma factors are essential bacterial proteins that bind to RNA polymerase (RNAP) to form the holoenzyme, enabling it to recognize specific promoter regions (-10 and -35 sequences) and initiate transcription. They regulate gene expression by guiding RNAP to specific genes, allowing rapid adaptation to environmental changes, such as stress or nutrient limitation.
What are the main cis elements of prokaryotic promoters and their respective functions that makes it different from eukaryotic promoters?
The main cis elements of prokaryotic promoters are the -10 element (Pribnow Box) and the -35 element. The -10 element (Pribnow box), with the consensus sequence TATAAT is essential for binding RNA polymerase (via the sigma factor) and facilitating DNA unwinding. Its AT-rich nature allows for easy separation of the DNA strands, crucial for initiation. The -35 element with the consensus sequence TTGACA has the primary function of serving as the initial binding site for the RNA polymerase holoenzyme, specifically recognized by the sigma factor subunit. It acts as an anchor to recruit RNA polymerase to the DNA, determining the strength of the promoter.
What are the main cis elements of eukaryotic promoters and their function that makes it different from prokaryotic promoters?
The main cis elements of eukaryotic promoters are the TATA Box (Hogness Box), Inr, the CAAT box, the GC Box. enhancers/silencers. The TATA box is a conserved DNA sequence (TATAAA) found in the promoter region of eukaryotic and archaeal genes, typically located 25–35 base pairs upstream of the transcription start site. The TATA box serves as the initiation site for transcription. It binds the TATA-binding protein (TBP), part of the transcription factor complex (TFIID), which subsequently recruits RNA polymerase to begin synthesizing RNA from DNA. The Initiator (Inr) element is a crucial core promoter DNA sequence, typically spanning 2–17 bp around the transcription start site (+1), that directs RNA polymerase II to begin transcription. TFIID, a basal transcription factor, recognizes and binds to the Inr via subunits TAF1 and TAF2. This binding forms the preinitiation complex (PIC). The CAAT box is a conserved eukaryotic promoter element located roughly -75 of the transcription start site. The CAAT box is a promoter element that binds transcription factors (such as CTF or C/EBP) to facilitate the initiation of transcription. The GC box is a distinct, GC-rich nucleotide pattern (consensus sequence often 5'-GGGCGG-3') found usually upstream of the TATA box. It binds the transcription factor Sp1, which helps initiate transcription. Enhancers and silencers are cis-regulatory DNA sequences that bind transcription factors to increase (enhancers) or decrease (silencers) gene expression. Enhancers bind specific transcription factors (activators) that recruit co-activators and RNA polymerase II to the promoter, forming an "enhanceosome". Silencers bind repressor proteins that prevent RNA polymerase from binding to the promoter or induce chromatin to close, making it inaccessible.
What is the core idea behind a luciferase reporter assay?
A luciferase reporter assay tells you how much a DNA sequence or protein condition changes transcription of a reporter gene. More luciferase signal means that there is more transcriptional activation of the reporter, and less luciferase signal means that there is less transcription, or repression.
What is the first general step in interpreting luciferase reporter data properly?
Step 1: Always ask what was changed
Before interpreting any graph, identify what the experiment manipulated:
Was the variable:
a DNA sequence?
-> then the assay is testing cis elements
a protein or a protein mutant/domain deletion?
-> then the assay is testing trans-acting protein function
both?
-> then it is testing whether a protein acts through a specific DNA element
What is the second general step in interpreting luciferase reporter data properly?
Step 2: Read the axes
Usually:
x-axis = constructs or conditions
y-axis = luciferase activity
often shown as:
relative luciferase units
fold activation
normalized luciferase activity
Higher bar = stronger reporter transcription.
What is the third general step in interpreting luciferase reporter data properly?
Step 3: Know the baseline
Interpretation depends on what the construct is compared to.
Common baselines:
empty vector
minimal promoter alone
wild-type promoter
untreated control
reporter without enhancer
wild-type protein
You always interpret relative to the control.
What is the fourth general step in interpreting luciferase reporter data properly?
Step 4: Use the basic interpretation rule
If reporter activity goes up
That usually means the tested sequence/protein has a positive effect on transcription.
If reporter activity goes down
That usually means the tested sequence/protein has a negative effect, or that an important activating function was lost.
What are 4 important patterns to memorize when it comes to interpreting luciferase reporter assay data?
Pattern 1
Mutation lowers activity
-> that sequence/domain is likely needed for activation
Pattern 2
Mutation raises activity
-> that sequence/domain is likely involved in repression
Pattern 3
Deleting a region lowers activity
-> deleted region likely contains a positive element/domain
Pattern 4
Deleting a region raises activity
-> deleted region likely contains a negative element/domain
What are some cis elements that can analyzed using luciferase reporter assays?
Cis elements are DNA sequences in the reporter construct, such as:
core promoter
TATA box
Initiator (Inr)
proximal promoter elements
enhancers
silencers
specific transcription factor binding sites
What are some typical protein manipulations done when testing protein domains using luciferase reporter data?
full-length protein
deletion mutants
point mutants
domain fusions
This tells you which protein domains are needed for activation or repression.
What are three reliable writing templates for interpreting luciferase reporter assay data of cis elements?
“Mutation/deletion of this region reduced reporter activity, indicating that this sequence contains a positive cis-regulatory element required for transcription.”
“Mutation of this motif increased reporter activity, suggesting that it functions as a negative cis-regulatory element.”
“The Inr mutation reduced basal luciferase expression, indicating that the Initiator contributes to core promoter function and transcription initiation.”
What are three reliable writing templates for interpreting luciferase reporter assay data of protein domains?
“Deletion of this domain abolished reporter activation, indicating that the domain is required for transcriptional activation.”
“Loss of repression after deletion suggests that the removed region contains a functional repression domain.”
“The mutant retained the DNA-binding region but lost activation, suggesting that the deleted region is needed for co-activator recruitment rather than DNA binding.”
Describe the stepwise process of 5’ capping in eukaryotes
In eukaryotes, 5′ capping is a co-transcriptional RNA-processing event that occurs very early, usually when the nascent pre-mRNA is about 20–30 nucleotides long.
1. Nascent RNA emerges from RNA polymerase II
The new transcript initially has a 5′ triphosphate end
The C-terminal domain (CTD) of RNA polymerase II helps recruit the capping machinery.
2. RNA 5′-triphosphatase acts
removes the terminal γ-phosphate from the 5′ end of the RNA.
Now the RNA has a 5′ diphosphate end.
3. Guanylyltransferase adds GMP to the 5′ diphosphate end of the RNA.
This forms an unusual 5′–5′ triphosphate linkage
This added guanine is the cap guanosine.
4. Guanine-N7 methyltransferase methylates the cap
Methyl donor: SAM (S-adenosylmethionine)
methylates the N7 position of the added guanine
This produces the 7-methylguanosine cap
This is the basic cap 0 structure.
What are the main players involved in 5’ capping in eukaryotes?
RNA polymerase II CTD: recruits capping enzymes
RNA 5′-triphosphatase: removes γ-phosphate
Guanylyltransferase: adds GMP
Guanine-N7 methyltransferase: makes m⁷G
2′-O-methyltransferases: make cap 1 and sometimes cap 2
SAM: methyl donor for methylation reactions
What are the main modifications in 5’ capping in eukaryotes?
1.) Removal of γ-phosphate
2.) Addition of GMP
3.) Formation of a 5′–5′ triphosphate linkage
4.) N7 methylation of guanine -> m⁷G cap
5.) Possible 2′-O-methylation of the first and second nucleotides
What is constitutive splicing?
Constitutive splicing is the default splicing pattern in which all introns are removed and all exons are joined together in the same way every time.
The same mature mRNA is produced from that pre-mRNA.
No exon choices are changed.
What is alternative splicing?
Alternative splicing is when the same pre-mRNA can be spliced in different ways, so different combinations of exons are included or excluded.
This allows one gene to produce multiple different mRNA isoforms
Those different mRNAs can produce different protein isoforms
What is the difference between constitutive splicing and alternative splicing?
Constitutive splicing always processes a pre-mRNA the same way, whereas alternative splicing allows different mature mRNAs to be made from the same gene.
What is RNA editing and what can it do?
The alteration of the sequence of nucleotides in the RNA after it has been transcribed from DNA but before it is translated into protein.
What it can do:
change one nucleotide to another
insert nucleotides
delete nucleotides
What is substitution editing and a simple example of it?
It is the chemical alteration of individual nucleotides (functionally the equivalent of point mutations but not considered a mutation because the gene is not changed).
Example: Can convert coding triplet to a stop codon to truncate protein.
What kind of editing is mammalian apolipoprotein B Glu codon conversion into a stop codon?
This is substitution editing, specifically:
C-to-U RNA editing
A cytidine in the RNA is chemically deaminated to uridine.
This is a post-transcriptional modification:
DNA is unchanged
RNA sequence is changed
protein output changes
What is the actual editing event that takes place during mammalian apolipoprotein B Glu codon conversion into a stop codon?
At a specific position in apoB mRNA:
original codon: CAA = glutamine
edited codon: UAA = stop codon
This happens because one cytidine (C) in the RNA is converted to uridine (U).
So: CAA -> UAA
That single base change creates a translation stop signal.
What are the functional consequences of the two proteins involved in mammalian apolipoprotein B Glu codon conversion into a stop codon (ApoB-100 and ApoB-48)?
ApoB-100
made in the liver
full-length protein
important for VLDL/LDL metabolism
contains the LDL receptor-binding region in the C-terminal part
ApoB-48
made in the intestine
shorter protein because translation stops early
used in chylomicron assembly
lacks the C-terminal portion of apoB-100
What are the players involved in the substitution editing of mammalian apolipoprotein B Glu codon conversion into a stop codon?
1.) APOBEC-1
2.) ACF / A1CF (APOBEC-1 complementation factor)
3.) The apoB RNA editing site
4.) Cis-acting RNA sequences
5.) The editosome
What is the role of APOBEC-1 in the substitution editing of mammalian apolipoprotein B Glu codon conversion into a stop codon?
This is the key catalytic enzyme.
Role
It is a cytidine deaminase
It converts C -> U in the RNA
What it does chemically
It removes an amino group from cytidine, converting it into uridine.
Importance
Without APOBEC-1, the editing reaction does not occur.
Why is the mammalian apolipoprotein B Glu codon conversion into a stop codon a classic example of substitution editing?
The apoB case is one of the most famous examples because it clearly shows that:
RNA can be changed after transcription
a single nucleotide substitution can drastically alter protein output
editing can be regulated by tissue
the edited RNA can produce a functionally different protein
What kind of RNA editing is glutamate receptor channel protein Gln codon converted to an Arg codon?
A to I substitution editing:
Adenosine Deaminase Acting on RNA (ADAR) enzymes, which convert adenosine residues to inosine in an mRNA molecule by hydrolytic deamination.
Why does the A to I substitution editing of glutamate receptor channel protein Gln codon converted to an Arg codon matters?
The Q/R site lies in the region of the receptor that helps form the ion channel pore.
That means the amino acid at this position strongly affects what ions can pass through the channel.
If the site is unedited: Q
the channel is more Ca²⁺ permeable
the channel tends to have higher conductance
the receptor is more susceptible to Ca²⁺ entry
If the site is edited: R
the positively charged arginine alters pore properties
Ca²⁺ permeability is strongly reduced
channel behavior changes in a protective way for neurons
So the key biological effect is:
Q → R editing makes the AMPA receptor less permeable to Ca²⁺
What is the common chemical mechanism behind substitution editing of A or C Residues?
Most common chemical mechanism: deamination
Deamination means removal of an amino group (-NH2) from a nitrogenous base.
This chemical change alters the identity/base-pairing properties of that nucleotide.
What are two common examples of deanimation in RNA editing by substitution?
1. A-to-I editing
enzyme removes the amino group from adenosine
adenosine becomes inosine
inosine is interpreted like guanosine (G)
So functionally: A -> I = G
2. C-to-U editing
enzyme removes the amino group from cytidine
cytidine becomes uridine
So: C -> U
What modification changes results from editing by substitution involving deanimation?
Because the base is chemically altered, the edited RNA may now have different:
codons
base-pairing behavior
splicing patterns
stability
protein-coding potential
For example:
a codon may specify a different amino acid
or it may become a stop codon
What is the general concept behind RNA editing via substitution for both C->U and A->I editing?
Both usually happen through a deamination-type modification, meaning the base is chemically altered so it behaves like a different base.
For both types, the logic is:
A specific RNA is made from DNA
An editing enzyme recognizes a particular site on that RNA
The enzyme changes one base into another base-like form
That edited base is then interpreted differently during:
translation
splicing
RNA folding
RNA function
What is the main concept behind C->U editing and a classic example of it?
In C-to-U editing, a cytidine in RNA is converted into uridine.
This can change:
a codon
the amino acid sequence
or even create a stop codon
A classic example is apoB mRNA editing, where CAA becomes UAA.
What are the main players involved in C->U editing?
1. Target RNA
The RNA that contains the cytidine to be edited
It must contain the correct nearby cis-acting recognition sequences
2. APOBEC family editing enzyme
The classic catalytic enzyme is APOBEC-1
APOBEC proteins are cytidine deaminases
Their role is to convert C → U
3. Accessory RNA-binding proteins
These help APOBEC edit the correct site
A classic example is A1CF/ACF in the apoB system
Other accessory factors can also help depending on context
4. Cis-acting RNA elements
These are nearby RNA sequences that help the enzyme recognize the proper editing site
In apoB, an important example is the mooring sequence
5. Editosome
The functional RNA-protein complex that performs editing
Includes the catalytic enzyme plus accessory factors
What do the modification in C->U editing means functionally?
Once the cytidine becomes uridine:
the codon may now code for a different amino acid
or may become a stop codon
so the final protein product can change dramatically
What is the main concept behind A -> I editing and what is a classic example of it?
In A-to-I editing, an adenosine in RNA is converted into inosine.
Inosine is important because the cell usually reads inosine like guanosine (G).
So functionally: A -> I = G
That means A-to-I editing can change:
codons
splice signals
RNA structure
miRNA targeting
RNA stability/function
A classic coding example is the GluA2 Q/R site.
What are the main players involved in A->I Editing
1. Target RNA
Usually a pre-mRNA or other RNA containing an editable adenosine
2. ADAR enzymes
The major enzymes are ADARs (adenosine deaminases acting on RNA)
Common members include ADAR1 and ADAR2
They catalyze the conversion A → I
3. Double-stranded RNA structure
ADARs usually do not edit single-stranded RNA sites efficiently on their own
They usually require the target adenosine to be in a double-stranded RNA region
4. Editing complementary sequence (ECS) or paired region
A nearby complementary RNA sequence often base-pairs with the target region
This forms the dsRNA structure recognized by ADAR
5. RNA-processing machinery
Because much A-to-I editing happens in pre-mRNA, splicing and nuclear RNA processing context matter
6. Ribosome / spliceosome / other readers
These cellular machines “interpret” inosine as if it were guanosine
That is how the edit affects protein sequence or RNA processing
What do the modifications in A->I editing means functionally?
Because inosine behaves like G:
a codon can change to encode a different amino acid
splice-site choice can change
RNA pairing properties can change
regulatory interactions can change
What is a snRNP?
A snRNP (small nuclear ribonucleoprotein) is a complex made of:
a small nuclear RNA (snRNA)
associated proteins
snRNPs are major components of the spliceosome, the machinery that removes introns from pre-mRNA.
What are the components of snRNP?
A snRNP contains:
1. snRNA
a small nuclear RNA such as U1, U2, U4, U5, or U6
helps recognize splice sites and participates in the splicing reaction
2. Proteins
common Sm or Sm-like proteins that help stabilize the snRNP
snRNP-specific proteins that give each snRNP its particular function
What are the functions of snRNPs?
snRNPs mainly function in pre-mRNA splicing.
They help:
recognize the 5′ splice site
recognize the branch point
bring splice sites together
form the spliceosome
catalyze intron removal and exon ligation
What does splicing do?
Splicing removes an intron from a pre-mRNA and joins the two surrounding exons together.
It is carried out by the spliceosome, a large RNA-protein complex built from snRNPs and other splicing factors.
What are the main cis elements in the pre-mRNA involved in splicing? (Textbook + ChatGPT)
5′ splice site (donor site/Splice donor)
Located at the 5′ end of the intron
end of the first exon
Usually begins with GU
Marks where the intron starts
Branch point sequence/site
Located (within) inside the intron, upstream of the 3′ splice site
Contains a critical branch-point adenosine (A)
This A performs the first nucleophilic attack
Polypyrimidine tract
Usually between the branch point and the 3′ splice site
Py rich upstream of the splice acceptor
Rich in U and C
Helps recruit key factors
3′ splice site (acceptor site/splice acceptor)
Located at the 3′ end of the intron
beginning of second exon
Usually ends with AG
Marks where the intron ends
Give an overview of the pre-mRNA splicing reaction with the general steps
Pre-mRNA splicing occurs through two site-specific transesterification reactions that result in phosphodiester bond cleavage and ligation. The 5′ and 3′ splice sites consist of the conserved sequence elements shown; the Py tract in the intron is a string of pyrimidine residues.
General Steps
1.) The branch-point 2’-OH attacks the 5’ splice site.
2.) The 5’ splice site is now activated to attack the 3’ splice site.
3.) The intron is released from the spliced mRNA as a lariat
Give an overview of the steps involved in spliceosome assembly on pre-mRNAs involving base pairing to snRNAs
1.) U1 binds to the 5’ splice site; U2 binds to the branch point
2.) The U4-U6-U5 trimeric snRNP displaces U1 at the 5’ splice site, then U4 dissociates
3.) U6 and U2 catalyze attack of the branch point on the 5’ splice site
4.) The 5’ splice site attacks the 3’ splice site, completing the reaction
Notice that in step 2, U6 snRNA base-pairs near the 5′ exon binding site where U1 snRNA was formerly bound. As U4 dissociates, the U2-U6-U5 complex remains assembled on the pre-mRNA. U5 base-pairs to both sides of the splice junction to align the RNA for the splicing reaction, and U2 and U6 base-pair to each other.
Fill in the blank
Splicing occurs through formation of a ___ structure and is a 2-step ___ process.
lariat, transterification
What are the 2 steps in the 2 step Tranesterification process that splicing occurs through the formation of a lariat structure? (lecture)
Step 1.) Transesterification produces a new ester bond at 2 ́ hydroxyl position of the branch point A ribose -> 2 ́, 5 ́ phosphodiester bond formed, phosphodiester bond 3 ́ position is broken
Step 2.) The free 3 ́ hydroxyl can attack P at the 5 ́ end of exon splice acceptor site. Excised lariat intron is processed by debranching enzyme and degraded
What are the main trans-acting factors in the stepwise splicing process and their roles?
U1 snRNP
Recognizes and base-pairs with the 5′ splice site
SF1 / BBP (branch-point binding protein)
Initially recognizes the branch point
U2AF
A heterodimer:
U2AF65 binds the polypyrimidine tract
U2AF35 helps recognize the 3′ splice site AG
U2 snRNP
Replaces SF1 at the branch point
Binds near the branch point so that the A is bulged out
Prepares the branch-point A for reaction
U4/U6.U5 tri-snRNP
A preassembled complex containing:
U4 snRNP
U6 snRNP
U5 snRNP
Roles:
U4: keeps U6 inactive at first
U6: becomes part of the catalytic center
U5: helps align the exons for ligation
SR proteins and other regulatory proteins
Help define splice sites
Promote proper spliceosome assembly
Important especially in exon recognition and alternative splicing
What are the steps in the stepwise spliceosome assembly?
Step 1: Early (E) complex formation
This is the first recognition stage.
What binds:
U1 snRNP binds the 5′ splice site
SF1/BBP binds the branch point
U2AF65 binds the polypyrimidine tract
U2AF35 binds the 3′ splice site
Purpose:
Marks the intron boundaries
Commits the pre-mRNA to splicing
Brings the key cis elements into an organized framework
Step 2: A complex (pre-spliceosome) formation
What happens:
U2 snRNP is recruited to the branch point
U2 replaces SF1/BBP
Important feature:
U2 binds in a way that leaves the branch-point A unpaired/bulged out
Purpose:
Positions the branch-point A for the first reaction
Step 3: B complex formation
What happens:
The U4/U6.U5 tri-snRNP joins the complex
Now the spliceosome contains: U1, U2, U4, U5, U6
Purpose:
Brings in the snRNPs needed to build the active catalytic spliceosome
Step 4: Spliceosome activation
This involves major rearrangements.
What happens:
U1 leaves the 5′ splice site
U6 replaces U1 at the 5′ splice site
U4 leaves, allowing U6 to become active
U2 and U6 interact extensively and form the catalytic core
U5 contacts the exon ends and helps align them
Important concept:
The spliceosome’s catalytic center is largely RNA-based, especially involving U2 and U6 snRNAs.
Purpose:
Convert the spliceosome into its active catalytic form
What is an important note for the chemical steps of the two transesterification reactions involved in splicing?
No net ATP is required for the chemistry itself
ATP is used for assembly and rearrangements, not for the transesterification reactions directly
Summarize the role of each major U sRNP
U1
binds the 5′ splice site
starts splice-site recognition
U2
binds the branch point
positions the branch-point A for catalysis
U4
keeps U6 inactive before activation
U6
replaces U1 at the 5′ splice site
forms the catalytic center with U2
U5
aligns the exon ends for exon ligation
What happens during the first transesterification reaction of splicing? (chatgpt)
Reaction:
The 2′-OH of the branch-point A attacks the phosphate at the 5′ splice site
Result:
The bond between exon 1 and the intron is broken
The 5′ end of the intron is joined to the branch-point A
This creates a lariat intron
Exon 1 is left with a free 3′-OH
Product after reaction 1:
free 5′ exon with a 3′-OH
lariat intron–3′ exon intermediate
Role of cis elements here:
5′ splice site = site of cleavage
branch-point A = nucleophile
U2 positions the branch A
U6/U2 help form the active center
What happens during the second transesterification reaction of splicing? (chatgpt)
Reaction:
The free 3′-OH of exon 1 attacks the phosphate at the 3′ splice site
Result:
Exon 1 and exon 2 are ligated together
The intron lariat is released
Product after reaction 2:
spliced mRNA exon-exon product
released lariat intron
Role of factors here:
U5 snRNP helps align the two exons so ligation can occur accurately
3′ splice site AG is the cleavage/ligation point
What is the Exon Definition concept?
Exon definition is the idea that, in many eukaryotic pre-mRNAs—especially in vertebrates with long introns and short exons—the splicing machinery first recognizes an exon as a unit, rather than recognizing the whole intron first.
What is the core idea behind the Exon Definition concept?
The spliceosome initially identifies an exon by pairing:
the 3′ splice site at the upstream end of the exon with the 5′ splice site at the downstream end of the same exon
So the exon gets “marked” or defined before the final splicing reactions occur.
How does the Exon Definition work?
1. Factors bind around the exon
U2AF and related factors recognize the upstream 3′ splice site, polypyrimidine tract, and branch region
U1 snRNP binds the downstream 5′ splice site
These interactions occur across the exon.
2. The exon is recognized as a legitimate exon
If both splice sites are recognized well, the exon is “defined” as something to keep.
3. Rearrangement to catalytic spliceosome
After exon definition, the complex rearranges so the correct 5′ splice site of one exon is paired with the correct 3′ splice site of the next exon across the intron, allowing actual intron removal.
Why is the Exon Definition concept particularly important to eukaryotes?
In higher eukaryotes:
introns are often very long
exons are relatively short
So it is often easier for the cell to recognize the small exon unit first, instead of trying to identify an entire long intron all at once.
How does the exon definition concept guide splicing in a given tissue?
Exon definition is strongly influenced by tissue-specific RNA-binding proteins.
Key idea
Different tissues express different:
SR proteins
hnRNP proteins
other splicing regulators
These proteins bind cis-regulatory elements on the pre-mRNA, such as:
ESEs = exonic splicing enhancers
ESSs = exonic splicing silencers
ISEs = intronic splicing enhancers
ISSs = intronic splicing silencers
What do proteins like SR proteins and hnRNP proteins do in the concept of the Exon Definition?
If activators bind
They help recruit or stabilize:
U1
U2AF
other spliceosome components
This strengthens exon definition, so the exon is more likely to be included.
If repressors bind
They block splice-site recognition or prevent factor assembly.
This weakens exon definition, so the exon is more likely to be skipped.
How does the exon definition relate to tissue-specific outcome?
Because different tissues express different regulatory proteins, the same exon may be:
well defined and included in one tissue
poorly defined and skipped in another tissue
That is one major basis of alternative splicing.
Example logic
In tissue A, an SR protein binds an ESE → exon recognition is strong → exon included
In tissue B, a repressor binds an ESS → exon recognition is weak → exon skipped
So exon definition is the framework through which tissue-specific splicing regulators decide whether an exon is used.
How many times are micro RNA’s (miRs) cleaved before they become mature miR?
2 Times
Drosha cleavage in the nucleus
Dicer cleavage in the cytoplasm
What is the first step of RNA cleaving in the process of microRNA synthesis?
Step 1: Transcription of the miRNA gene
RNA involved
pri-miRNA (primary miRNA transcript)
Proteins involved
usually RNA polymerase II
What is produced
a long pri-miRNA
contains a stem-loop / hairpin structure
may also have a 5′ cap and poly(A) tail if transcribed by Pol II
Purpose
This is the original precursor RNA that will be processed into a mature miRNA.
What is the second step of RNA cleaving in the process of microRNA synthesis?
Step 2: Nuclear cleavage by the Microprocessor complex
This is the first RNA-cleavage step.
RNA involved
pri-miRNA
Proteins involved
Drosha
an RNase III enzyme
performs the actual cleavage
DGCR8 (called Pasha in some organisms)
an RNA-binding partner of Drosha
recognizes the pri-miRNA hairpin and helps position Drosha correctly
Complex involved
Microprocessor complex = Drosha + DGCR8
What happens
DGCR8 recognizes the hairpin region in the pri-miRNA
Drosha cleaves near the base of the hairpin
this releases a shorter hairpin called the pre-miRNA
Product
pre-miRNA
typically a ~70 nt hairpin with a 2-nt 3′ overhang
Purpose
This cleavage excises the hairpin precursor from the larger pri-miRNA transcript.
What is the third step of RNA cleaving in the process of microRNA synthesis?
Step 3: Export from nucleus to cytoplasm
RNA involved
pre-miRNA
Proteins involved
Exportin-5
nuclear export receptor
recognizes the pre-miRNA hairpin and its 3′ overhang
Ran-GTP
provides energy/directionality for export
What happens
Exportin-5 binds the pre-miRNA
exports it through the nuclear pore into the cytoplasm
Purpose
Moves the pre-miRNA to the cytoplasm for the next cleavage step
What is the fourth step of RNA cleaving in the process of microRNA synthesis?
Step 4: Cytoplasmic cleavage by Dicer
This is the second RNA-cleavage step.
RNA involved
pre-miRNA
Proteins involved
Dicer
another RNase III enzyme
cleaves the loop region of the pre-miRNA hairpin
Accessory proteins
Depending on the organism/cell type, Dicer often works with proteins such as:
TRBP (TAR RNA-binding protein)
PACT
These help Dicer processing and loading into Argonaute.
What happens
Dicer measures from the pre-miRNA end
cleaves the hairpin near the loop
generates a short miRNA duplex
Product
miRNA duplex
about ~21–23 nucleotides
contains:
guide strand
passenger strand
usually has 2-nt 3′ overhangs
Purpose
Produces the short RNA duplex from which the mature miRNA will be selected.
What is the fifth step of RNA cleaving in the process of microRNA synthesis?
Step 5: Loading into Argonaute / RISC assembly
RNA involved
miRNA duplex
Proteins involved
Argonaute (AGO)
core protein of the RISC complex
binds the miRNA and uses it to recognize target mRNAs
RISC-loading machinery
includes Argonaute and associated factors
What happens
the miRNA duplex is loaded into Argonaute
one strand is selected as the guide strand
the other strand is the passenger strand (miRNA*)
Fate of the passenger strand
usually removed and degraded
in some cases may be cleaved or unwound, depending on complementarity and AGO type
Product
mature miRNA-loaded RISC
Purpose
Creates the active silencing complex that can regulate target mRNAs.
What is the sixth step of RNA cleaving in the process of microRNA synthesis?
Step 6: Mature miRNA function
RNA involved
mature guide miRNA
target mRNA
Protein involved
Argonaute
What happens
the miRNA guides Argonaute to target mRNAs by base pairing
this leads to:
translational repression
mRNA destabilization/deadenylation
sometimes mRNA cleavage if complementarity is very high
What are the main RNAs involved in the steps of RNA cleaving in the process of microRNA synthesis?
pri-miRNA = primary transcript
pre-miRNA = Drosha-cleaved hairpin precursor
miRNA duplex = Dicer product
mature miRNA = guide strand in Argonaute
target mRNA = RNA regulated by the mature miRNA
What are the main proteins involved in the steps of RNA cleaving in the process of microRNA synthesis?
RNA polymerase II = transcribes pri-miRNA
Drosha = nuclear RNase III cleavage
DGCR8 = pri-miRNA recognition partner
Exportin-5 = exports pre-miRNA
Ran-GTP = nuclear export factor
Dicer = cytoplasmic RNase III cleavage
TRBP / PACT = Dicer-associated factors
Argonaute (AGO) = effector protein of RISC
miRNAs can reduce gene expression in what two related ways?
1.) translation blocking (translational repression)
2.) mRNA degradation / decay
Both are guided by the same basic principle: a miRNA in Argonaute/RISC base-pairs with a target mRNA, usually in the 3′ UTR.
What is similar between miRNA (miR) blocking and mRNA degradation as controlled by miRs?
Both mechanisms:
are directed by a miRNA
use the RISC/Argonaute complex
require base pairing between the miRNA and target mRNA
reduce the amount of protein produced
usually occur after transcription, so they are post-transcriptional regulation