Neurogenetic Disorders
1/47
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
48 Terms
What gene is mutated in Rett syndrome?
MECP2 gene on the X chromosome.

What is the inheritance pattern of Rett syndrome?
X-linked dominant, typically lethal in males.

Why are males rarely affected in Rett syndrome?
MECP2 mutations are lethal in hemizygous males, leading to miscarriage.

At what age does developmental regression begin in Rett syndrome?
Between 6–18 months.

What are hallmark stereotypic movements in Rett syndrome?
Hand-wringing, hand-washing, clapping/tapping.

What causes acquired microcephaly in Rett syndrome?
Slowing of head growth due to impaired neuronal maturation.

What neurologic symptoms occur in Rett syndrome?
Seizures, irregular breathing, loss of speech(Developmental regression), gait abnormalities, hand washing movements!

What is the average life expectancy in Rett syndrome?
Average ~24 years, but many survive into 40s–50s.

What is the inheritance pattern of Fragile X syndrome?
X-linked disorder (variable expression due to X-inactivation in females).

What gene is mutated in Fragile X syndrome?
FMR1 gene with CGG trinucleotide repeat expansion.

What protein is deficient in Fragile X syndrome?
FMRP (Fragile X Mental Retardation Protein).
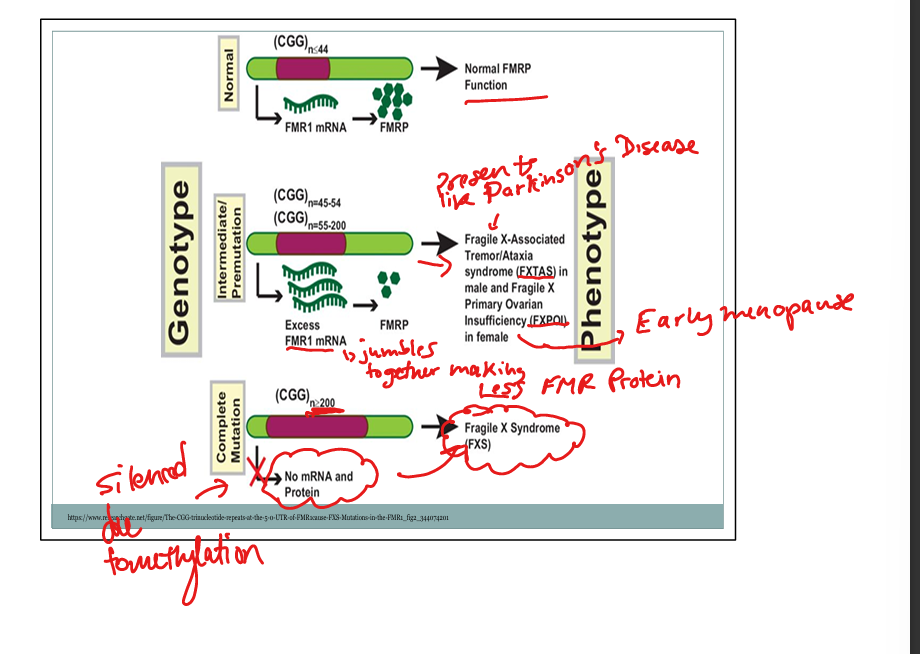
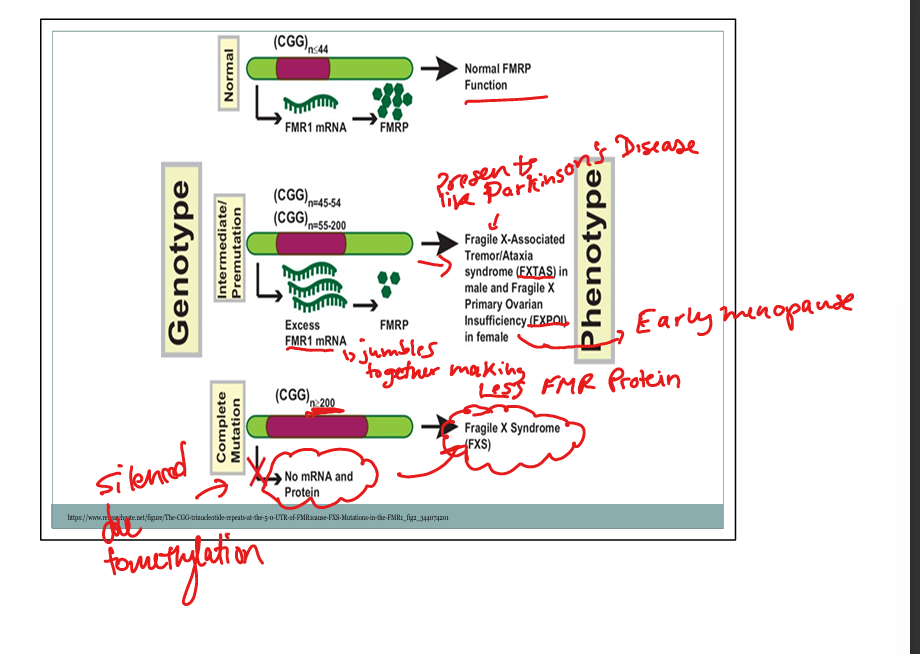
What is the mechanism of disease in Fragile X full mutation?
Hypermethylation → transcriptional silencing → loss of FMRP.
What are classic physical features of Fragile X in males?
Long face, large ears, macroorchidism(enlarged testicles in males) after puberty.

What neurodevelopmental disorders are associated with Fragile X?
Autism spectrum disorder, ADHD, intellectual disability.

What condition occurs in male premutation carriers? Females?
Males: Fragile X–associated tremor/ataxia syndrome (FXTAS). Females: Fragile X primary ovarian insufficiency (FXPOI).

What type of disorder are mucopolysaccharidoses (MPS)? Due to?
Lysosomal storage disorders due to GAG degradation enzyme deficiencies.

What accumulates in MPS disorders?
Glycosaminoglycans (GAGs).

What are common features of Mucopolysaccharides disorders?
Coarse facial features, joint contractures, short stature, hepatosplenomegaly, developmental delay.

What enzyme is deficient in Hunter syndrome?
Iduronate‑2‑sulfatase.

What is the inheritance pattern of Hunter syndrome?
X-linked recessive (“Hunter aims for the X”).

Does Hunter syndrome have corneal clouding?
No — vision is preserved.
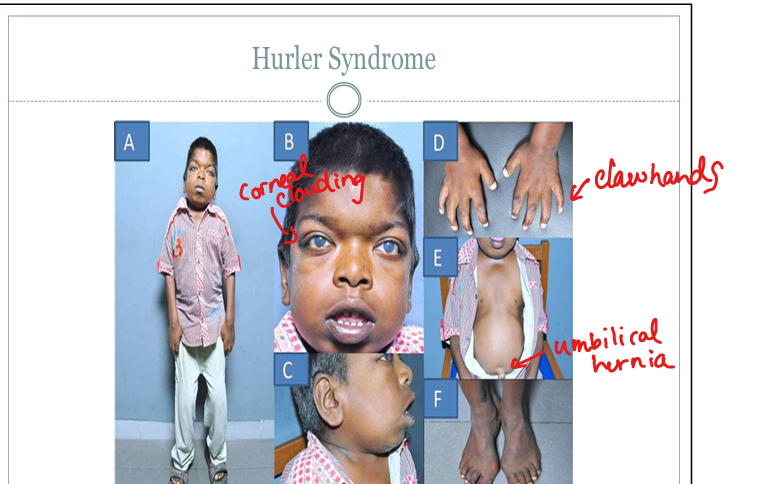
What enzyme is deficient in Hurler syndrome?
Alpha‑L‑iduronidase.

Does Hurler syndrome have corneal clouding?
Yes — corneal clouding is a hallmark.

What are shared features of Hunter and Hurler syndromes?
Coarse facies, claw hands, hepatosplenomegaly, developmental delay, macrocephaly.

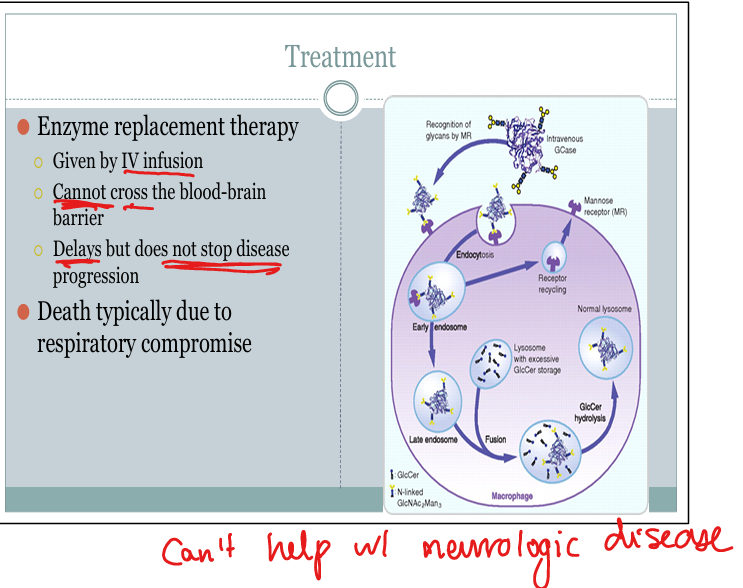
Why does enzyme replacement therapy not treat neurologic symptoms in MPS?
Enzymes cannot cross the blood–brain barrier.
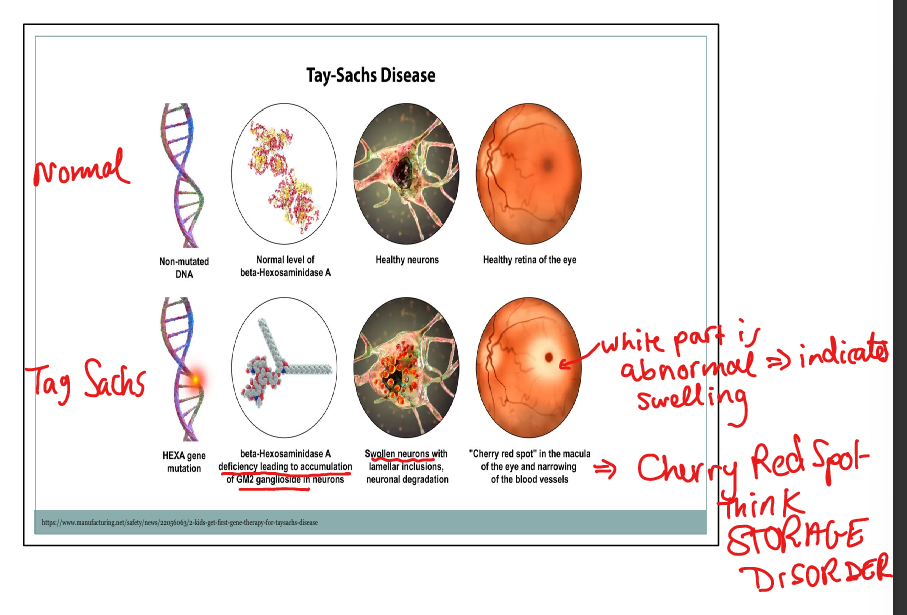
What gene is mutated in Tay‑Sachs disease?
HEXA gene encoding beta‑hexosaminidase A.

What is the inheritance pattern of Tay‑Sachs?
Autosomal recessive.

What population has high carrier frequency for Tay‑Sachs?
Ashkenazi Jewish population (1:30 carriers).

What substrate accumulates in Tay‑Sachs?
GM2 ganglioside.

What is the classic ophthalmologic finding in Tay‑Sachs?
Cherry‑red spot on the macula.
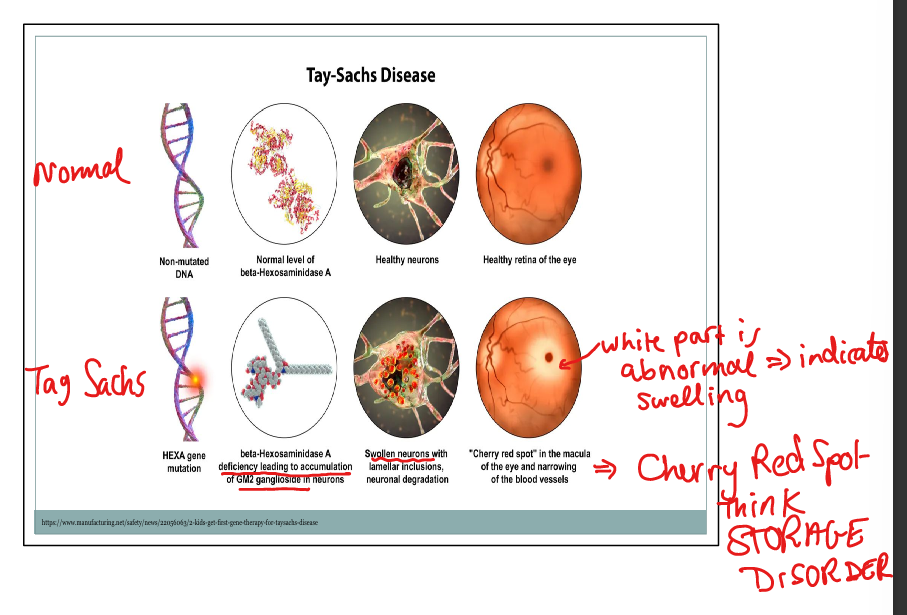
What causes the cherry‑red spot in Tay‑Sachs?
Surrounding retinal ganglion cell swelling makes the fovea appear red by contrast.
When do symptoms of Tay‑Sachs begin?
3–6 months of age.

What early symptoms occur in Tay‑Sachs?
Loss of motor skills, hypotonia, exaggerated startle response.

What later symptoms occur in Tay‑Sachs?
Seizures, blindness, progressive neurodegeneration.

What is the typical age of death in Tay‑Sachs?
2–5 years, usually from respiratory failure.

What is the inheritance pattern of Friedreich ataxia?
Autosomal recessive.

What gene is mutated in Friedreich ataxia?
FXN gene with GAA trinucleotide repeat expansion.

What protein is deficient in Friedreich ataxia? Location? Function?
Frataxin. Located in mitochondria. Iron binding and assembly of mitochondrial respiratory chain complexes I–III.

What tissues are most affected in Friedreich ataxia?
Cerebellum, dorsal root ganglia, corticospinal tracts, spinocerebellar tracts.

What are hallmark neurologic findings in Friedreich ataxia?
Ataxia, dysarthria(trouble speaking), loss of vibration/position sense, absent DTRs(hyporeflexia).

What systemic complications occur in Friedreich ataxia?
Hypertrophic cardiomyopathy, diabetes mellitus.

What is the typical age of onset in Friedreich ataxia?
10–15 years.

What is the typical cause of death in Friedreich ataxia?
Cardiomyopathy or respiratory infection.

What medication treats Friedreich ataxia?
Skyclarys (omaveloxolone). Activates Nrf2 to improve mitochondrial function and reduce oxidative stress.

Which disorders present with developmental regression?
Rett syndrome, Tay‑Sachs, Hurler syndrome, Hunter syndrome.
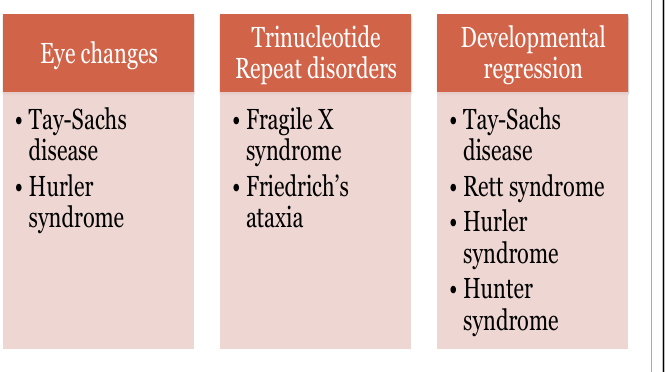
Which disorders show cherry‑red spots?
Tay‑Sachs (and other GM2 gangliosidoses).
Which disorders involve trinucleotide repeats?
Fragile X (CGG), Friedreich ataxia (GAA).
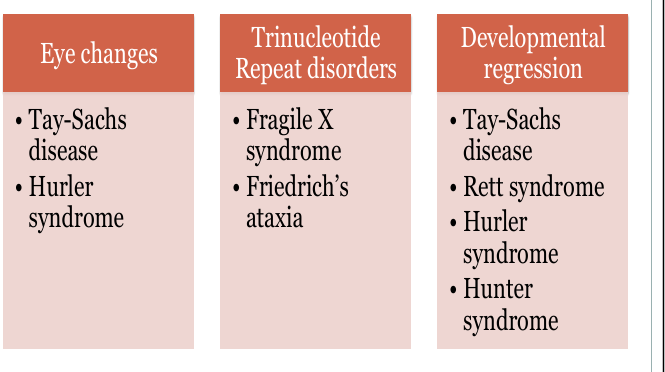
Which disorders are X‑linked?
Hunter syndrome (XLR), Rett syndrome (XLD), Fragile X (X‑linked).