GENE221 Mod 6 - Comparative microbial genomics
1/17
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
18 Terms
What are the benefits of using a metagenomics approach over 16S sequencing?
16S sequencing can give us an understanding of the taxes of microbes in a sample and populations within a sample, since it is an RNA sequence that is always present in bacteria/archaea. 18S sequencing is similar but with a distinctive sequence found in all fungi.
Metagenomics can give us far more data about the metabolic potential of a population and their genetic makeup i.e. how they relate to other organisms genetically.
What other benefits can metatranscriptomics offer compared to metagenomics?
Metatranscriptomics is done via Total RNA Sequencing (RNASeq). It can give the same information at genome sequencing but with other advantages.
Advantages:
We can determine gene expression of microbes within natural environments. We can see the diversity of active genes in a community, quantify gene expression under different conditions.
It can provide information about differences in the active functions of microbial communities which appear to be the same in terms of microbe composition
Can be used to study the virosphere! (but also all the parasites, bacteria, fungi = infectome) as well as the host gene expression
What types of analysis can you do with metatranscriptome data?
De novo assembly: putting short sequence reads together to reconstruct longer sequences (contigs). Contigs = contiguous sequences from shorter, overlapping reads. Annotating: once assembled, identifying where the contigs came from using sequence homology
Map sequence reads to a reference sequence
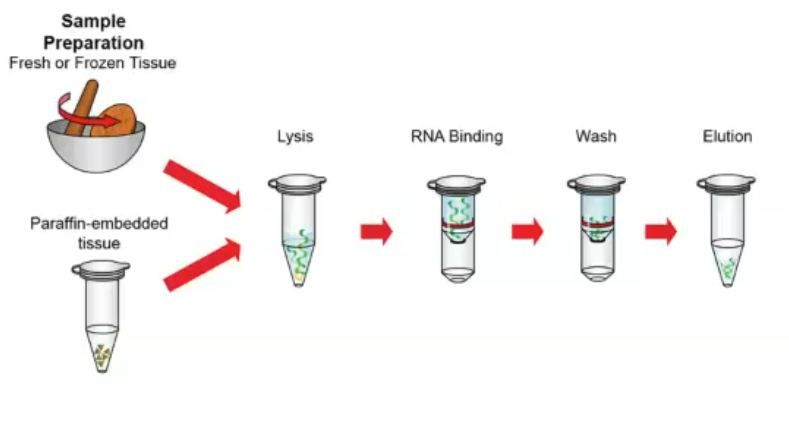
What causes for bias does metagenomics/metatranscriptomics have?
There is potential for sampling bias in data collection. The following points are important to think about.
Sequencing platforms – error rate, biases, read length, noise
• Coverage/depth
• Sample collection and preservation - contamination can overwhelm the real signal
DNA/RNA extraction methods
Nucleic acid extraction from all cell types to avoid bias
Should be effective for diverse microbial taxa
Very small quantities are usually sufficient for sequencing
• Enrichment or depletion steps can affect microbes.
• One sample is only representative of a single time point
What limitations does metagenomics/metatranscriptomics have?
Many genetic sequences are left not annotated because we don’t know what protein they encode. Our understanding of microbial communities is partial based on what we can infer from existing knowledge (i.e. what is well-characterised and exists in databases). We need better computational tools to deal with all the data we can now produce, which is where AI comes in.
Dark matter in metatranscriptomics makes up a vast portion of the transcript data we produce, but transcripts with no sequence homology to anything we understand so are left unannotated.
• What can artificial intelligence offer to metatranscriptomics?
The metagenomic identification of viruses is currently limited to those with sequence similarity to known viruses. Highly divergent viruses that comprise the “dark matter” of the virosphere remain challenging to detect. Over the past decade, artificial intelligence related approaches, especially deep learning algorithms (i.e. AlphaFold), have had a huge impact on protein structure prediction. Possibly AI could be used to help uncover more “dark matter”
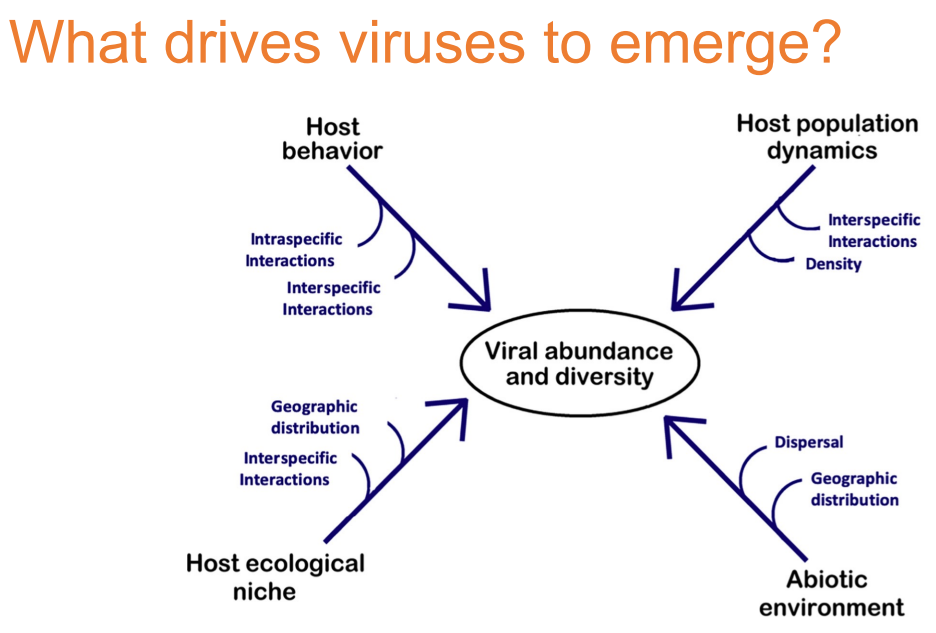
What factors seemingly shape the composition of the viral communities?
Virus emergence is often caused by the disruption or invasion of an ecological niche.
Abiotic and biotic factors have been identified that potentially modulate microbial community diversity and structure.
Identifying factors that promote diversification is crucial to understanding disease and its emergence. For example, host ecology and behavior affect contact rates among hosts, and are therefore likely to be important in shaping viral dynamics
What is preferential host switching and why is it important for understanding virus virulence and emergence?
Viruses are more easily transmitted between closely related hosts. If exhibited in real-world conditions, preferential host switching would mean that host taxonomy plays a key role in shaping virome composition.
What is the most studied factor studied for microbial diversity in humans?
Host age is arguably the most studied demographic factor that influences virome composition.This has been tested across a wide range of host species e.g. humans: Gut viral diversity is age-dependent
What are the 2 main abiotic factor that may shape microbial community diversity?
Ocean warming impacts on marine species distributions. As species ranges shift poleward in response to changing climates, individuals will be exposed to new microbes that differ in composition compared to those in their historical range
Host Biodiversity. Biodiversity increases closer to the equator. The dilution effect hypothesis posits a negative relationship between disease risk and host diversity. That is, high host diversity “dilutes” disease risk.
How might you go about testing whether these factors drive microbial composition in a range of host species?
An ecosystems approach to studying disease emergence. An ecosystem focus not only encompasses emerging and pathogenic viruses, but the entire virome in healthy and unhealthy hosts, as well as the complex interactions between viruses, hosts, and environments.
When sampling for a population or ecosystem:
Controls can be difficult to incorporate - Collect as much meta data as possible (sex, age, location, host, pH, temperature, etc.). Collect longitudinal (over time) data when possible
What is the use-case for genomics/transcriptomics rather than metagenomics for understanding viruses?
By individually sequencing genomes or transcriptomes in a population and comparing them, we can better understand:
Emergence events (time, source, geography)
Transmission chains
Evolution of variants
Spread through time and space
Outbreak’s trajectory
Changes in disease severity (virulence)
What data analysis techniques can we use to help find outbreak trajectory and evolution?
Establishing a phylogenetic tree of the virus which accounts for area of the world the virus is found and the time that it infects something, we can start to build a timeline and disease transmission chain and understand patterns of how a virus may spread and evolve with time and new enivronments.
Do pathogens become more or less virulent (severe) over time?
While classical theory states that pathogens/viruses would evolve to become less virulent, as viruses that were virulent enough to kill a large portion of their hosts would have worse transmission. This, coupled with hosts becoming more resistant to the virus, is a strong case for virus virulence decreasing over time. Unfortunately this is not often observed in nature, as ancient viruses still infect people today and many viruses continually split into more and more varied strains to fit any ecological niche.
What is the minimal genome concept?
A genome that contains only the genes required to sustain freeliving self-replication
How do many viruses optimize for the amount of information they carry in a small genome?
Many viruses will have overlapping reads or coding sequences. This is to maximise the amount of proteins they can synthesize with the same space in a genome. RNA viruses will contain very few non-coding RNA sequences. Even large DNA viruses will have most of the non-coding sequences important transcription signals like promoters, termination signals and functional RNA molecules.
What are essential genes?
Conditionally essential - Essential depending on what conditions are used for growth. Essential but redundant - Some genes can do the same essential function but deletion of either gene is tolerated
how do you test which genes are essential?
We will look at four approaches:
1. Look in nature for the smallest genomes
2. Compare distantly related genomes computationally to identity conserved (essential?) genes
3. Take a small natural genome and mutate genes to find out which ones are not essential 4. Creating a synthetic organism!