In Silico Methods for Drug Discovery
1/25
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
26 Terms
What are ligand based drug designs?
Uses knowledge of ligand structure with or without knowledge of the receptor to create models based on structural properties able to discriminate good compounds from bad.
QSAR and pharmacophore screening
What are structure based drug designs?
Uses knowledge of the receptor structure to guide the design of new compounds able to exploit unfulfilled interactions and shape
complementarity
(target) Protein-ligand (drug) docking
What methods are there to measure or compute how compounds sit in the active site of an enzyme and what does it mean to be able to?
Measure - X-ray crystallography, neutron diffraction, solution NMR
Computing - Protein-ligand docking
Can make judgements on the relative affinities of the compounds (scoring and ranking)
Can suggest modifications to the compound that will increase its affinity.
What is necessary for protein ligand docking/virtual ligand screening?
The receptor structure
From experiment (X-ray crystallography, neutron diffraction, NMR)
From homology modelling (3D structure derived by modelling using a similar protein with known 3D structure as a template)
The location of the active site (binding cavity)
Often know since many protein X-ray crystal structures include a bound ligand.
If not known, then must use cavity-detecting software.
The structures of the compounds to be docked (drawn and geometry optimised - same process as generating compound structures for QSAR)
What is docking?
Computational methods for finding the best matching between two molecules, a receptor and a ligand. Needs:
A posing method - placing the ligand into the active site
A scoring method - to give each individual pose a score in order to determine the best pose (and later to rank a compound set)
What problems does the posing method come with and how is it overcome?
The protein and the ligand are both flexible so there’s hundreds of degrees of freedom and an impossibly large number of possible conformations
Compromise: fully flexible ligand; rigid protein with
flexible binding site residues.Dock ligand into binding pocket → generate a large number of possible orientations, score each one by an energy function and select the best set
What are the 4 posing methods?
Random searching: Genetic algorithms and Monte Carlo simulations
Fast shape matching
Incremental construction
Simulated annealing
Describe how Random searching: Genetic algorithms works?
A class of computational problem-solving approaches that adapt the principles of biological competition and population dynamics
Model parameters (ensemble of possible ligand conformations) are encoded in a ‘chromosome’ and stochastically varied. Chromosomes yield possible solutions to a given problem and are evaluated by a
fitness function.The chromosomes that correspond to the best intermediate solutions are subjected to crossover and mutation operations to produce the next generation
Describe how Random searching: Monte Carlo simulations (simplified) works?
Generate an initial configuration of a ligand in an active site consisting of a random conformation, translation and rotation.
Score the initial configuration.
Generate a new random configuration and score it.
If the new solution scores better than the previous one, it is accepted.
Repeat previous steps until the desired number of configurations is obtained
Describe how Fast shape matching works?
Match triangles of interaction sites onto complementary ligand atoms
Analyse receptor
Look at features good for binding (H bond donors and acceptors)
Look at flexibility in active site
Analyses test compound and does the same and places whats needed in the active site
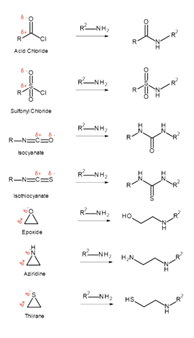
Describe how incremental construction works?
The receptor interaction surface is derived from crystallographic information and approximated by a finite set of interaction centers.
The ligand is fragmented into base fragments.
The ligand fragments are placed into the active site by matching the interaction centers.
The number of solutions is reduced by clash testing.
The base fragments are linked in compliance with a torsional database or a force field
What problems does the scoring method come with?
Ligand-binding events are driven by a combination of enthalpic
and entropic effects, either of which can dominate specific
interactions.
Most scoring functions are much more focused on capturing energetic (electrostatic & van der Waals interactions) than entropic effects
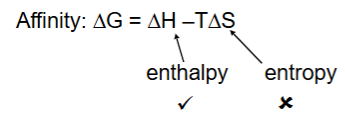
Which component of binding free energy do scoring functions model most accurately?
ΔH (enthalpy) - interactions like hydrogen bonds, ionic forces, and van der Waals contacts.
Why is ΔS difficult to calculate in docking?
Because entropy involves complex changes in ligand freedom, protein flexibility, and water displacement, which are hard to model computationally.
What happens to water molecules during ligand binding?
Water molecules in the binding site are displaced, increasing entropy and contributing favourably to binding.
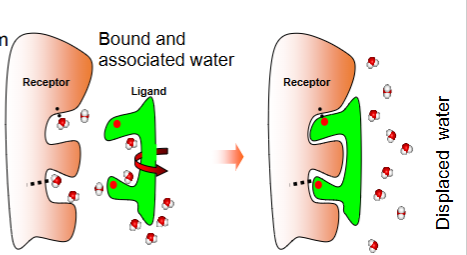
What entropic penalty occurs when a ligand binds to a protein?
Loss of ligand rotational and translational freedom.
What are the 3 major classes of scoring functions that are applied?
Force field based
Empirical
Knowledge based scoring functions
What is potential of mean force or knowledge based methods?
Description of observed interatomic distances and/or frequencies implying that these describe favorable/unfavorable interactions
Interaction potentials between each atom pair in 2 molecules (eg, ligand and protein) approximate the free energy of each pair-wise interaction as a function of inter-atomic distance.
For fast calculations the interaction energies for given pairs of atoms can be stored in a data table and retrieved quickly for calculating the PMF scores
Computational simplicity → permits efficient screening of large compound databases.
What is the disadvantage of potential of mean force or knowledge based methods?
Their derivation is essentially based on information implicitly encoded in limited sets of protein-ligand complexes
What are the limitations of potential of mean force or knowledge based methods?
Scores scale poorly with ligand molecular mass and the number of rotatable bonds.
Large molecules can form many hypothetical interactions in binding sites and therefore have the tendency to generate better scores than smaller compounds.
The entropy penalty for immobilisation of rotatable bonds, which is frequently not taken into account, scales with the number of such bonds.
Thus, if entropy penalties are included, flexible molecules tend to score lower than more rigid ones.
What are the complications that comes with protein ligand docking?
Being able to predict binding conformations and compound activity:
Limited resolution of crystallographic targets
Inherent flexibility of the protein
Induced fit or other conformational changes upon binding might not be seen
The participation of water in protein–ligand interactions
What are the computer approaches for receptor flexibility?
Generate an ensemble of receptor conformations (e.g. from NMR, X-ray, MD) and dock to those. A NMR protein structure (obtained in solution) may contain 10-20 conformations – use each of these to dock into
X-Ray crystal structures - X-ray crystal structure of the particular protein has been determined many times with a range of different ligands. The protein conformation will be slightly different in each case
Molecular dynamics
How is molecular dynamics done?
The force on each atom is calculated from a change in potential energy between current and new positions.
Atomic forces and masses are then used to determine atomic positions over series of very small time steps.
This provides a trajectory of changes in atomic positions over time.
Sample the protein’s conformations at various points in a
molecular dynamics trajectory and dock into these.
When is molecular dynamics done?
Run a molecular dynamics simulation after docking to examine the stability of the docked conformation and the strength of binding
Allows for receptor and ligand rearrangements to obtain lower energy conformations of the docked complex.
Computationally expensive (can take days for a single calculation)
What are the benefits and problems for molecular dynamics?
Benefits
Side-chain and backbone movements but restricted by simulation time
Can find novel conformation to limited extent
Moderate to high level of ligand effects
Quantification of conformational changes
Can handle solvent and ionic effects and membrane-bound proteins
Potential problems
High computational cost
Analysis of huge output effort-intensive
Biased by starting structure and initial velocity
May fail owing to short times of simulations and may miss best solution
What would you not want to include in a drug molecule?
Reactive functional groups
They increase the likelihood of toxicity
Decrease metabolic stability
Lead to non-specific binding to proteins or DNA, rather than just the desired therapeutic target