04. GLP, CSA/DEA
1/142
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
143 Terms
Which Act gives rise to GLPs and is the law that gives authority to the FDA to enforce them.
Federal Food, Drug, and Cosmetic ACT of 1938 - 21 CFR Part 58
WHAT ARE GLP?
Establish standards for conducting and reporting nonclinical safety testing
Allows for a quality product and accurate repetition of the study
Apply to nonclinical laboratory studies intended to support applications for research or marketing permits for products regulated by the FDA
it is a management system
Good Laboratory Practices
Regulates nonclinical laboratory studies
-Human and animal drugs
-Medical devices for humans
-Biological products (exclude hormones like insulin)
-Human food and color additives
-Animal food additives
-Electronic products (e.g. MRI)
What is a nonclinical laboratory study?
in vivo or in vitro experiments in which test articles are studies prospectively in test systems under laboratory conditions to determine their safety
What is not included in a nonclinical lab study?
-no studies using human subjects
-no clinical studies
-no field trials in animals
-no basic exploratory studies (utility, physical or chemical characteristics of test article
What does GCP stand for?
GCP = Good clinical practices
What does GLP stand for?
GLP = Good Laboratory Practices
What does GMP stand for?
GMP = Good manufacturing practices
What does IND stand for?
IND = Investigational new drug
What does NDA stand for?
NDA = new drug application
Definition of Testing Facility
person who actually conducts the study (i.e. uses the test article in the test system)
A "testing facility" can be a "person"
Definition of Person
individual, partnership, corporation, association, scientific or academic establishment, government agency, or any other legal entity
Definition of Nonclinical Laboratory Study
in vivo or in vitro experiments in which test articles are studied prospectively in test systems under laboratory conditions to determine their safety
Does not include: basic exploratory studies, studies utilizing human subjects or clinical studies, or field studies in animals
Definition of Sponsor**
Sponsor:
(1) A person who initiates and supports a nonclinical lab study; or
(2) A person who submits a nonclinical study to the FDA in support of an application for a research or marketing permit; or
(3) a testing facility, if it both initiates and conducts the study
Definition of Test system
animal, plant, microorganism, or subparts thereof to which a test or control article is administered or added for study. Includes appropriate groups or components of the system not treated with the test or control articles
Definition of Test article
any food or color additive, drug, biological product, electronic product, medical device for human use or any other article subject to regulations
Definition of study Director (SD)
individual with overall responsibility of the study
includes: technical conduct of the study; interpretation, analysis, documentation, and reporting of results;
represents the single point of study control
The study director shall assure what 6 things?
- The protocol, including any change, is approved and is followed.
- All experimental data are accurately recorded and verified.
- Unforeseen circumstances that may affect the quality and integrity of the study are noted when they occur, and corrective action is taken and documented.
- Test systems are as specified in the protocol.
- All applicable good laboratory practice regulations are followed.
- All raw data, documentation, protocols, specimens, and final reports are transferred to the archives during or at the close of the study.
Definition of Study Initiation date
the date the protocol is signed by the SD
Definition of Study Completion date
the date the final report is signed by the SD
Application for research or marketing permit includes what 8 things?
(1) An investigational new drug application (IND).
(2) A new drug application (NDA).
(3) A Notice of Claimed Investigational Exemption for a New Animal Drug.
(4) A new animal drug application.
(5) An application for a biologics license.
(6) An application for an investigational device.
(7) An Application for Premarket Approval of a Medical Device.
(8) A Product Development Protocol for a Medical Device.
Each individual engaged in the conduct of or responsible for the supervision of a study shall have ____, to enable that individual to perform the assigned functions.
education, training, and experience, or combination thereof
Each testing facility shall maintain _____ for each individual engaged in or supervising the conduct of a study.
a current summary of training and experience and job description
How many people need to be available for the study to be conducted?
a sufficient number to conduct the procedure according to protocol
____ personnel should not work with animals (or test or control articles) if it will affect study.
Ill
For each nonclinical laboratory study, testing facility management shall:
Designate a ____
Replace the ___ promptly if it becomes necessary
Assure there is a ____
Assure test and control articles are tested for ID, strength, purity, stability and uniformity
Assure personnel, resources, facilities, equiment, materials and methodologies are available as scheduled
Assure that personnel ____ functions they are to perform
Assure than any deviation from these regulations are reported by the ____ to the ____ and corrective actions are taken and documented
Study director
study director
Quality assurance unit (QAU)
e
e
understand
QAU to the Study Director
Definition of Quality Assurance Unit (QAU)
Responsible for monitoring each study to assure management that the facilities, equipment, personnel, methods, practices, records, and controls are in conformance with the regulations in this part
If someone is apart of the Quality Assurance Unit (QAU) can they also be involved in the study?
No, must be entirely separate from and independent of the personnel engaged in the direction and conduct of that study.
Who must QUALITY ASSURANCE UNIT(QAU) report deviations to?
Report deviations to the study director
The QAU is responsible for maintaining records of what?
1.Master schedule (indexed by test article)
2.Copy of protocols
3.Inspections (Dates, what was inspected, name of inspector, findings, actions recommended and actions taken)*.
*FDA inspections do not require access to these
The QAU is like the ____ and the study director is like the ____
IACUC
Insitutional Official
QAU is alot like the IACUC - so what shall they do?
Copy of master schedule sheet of all NCL studies at testing facility – include test article and containing the test system, nature of study, date study was initiated, current status of each study, identity of the sponsor, and name of the study director
Maintain copy of all protocols
Inspect each nonclinical laboratory study at intervals – include: maintain written and properly signed records of each periodic inspection showing the date of the inspection, the study inspected, the phase or segment of the study inspected, the person performing the inspection, findings and problems, action recommended and taken to resolve existing problems, and any scheduled date for reinspection.
Any problems found that affect study integrity – report to study director immediately
management and the study director written status reports on each study – note problems & corrective actions
no deviations from approved protocols or standard operating procedures were made without proper authorization and documentation
Review the final study report to assure that such report accurately describes the methods and standard operating procedures, and that the reported results accurately reflect the raw data of the NCL study
Prepare and sign a statement to be included with the final study report which shall specify the dates inspections were made and findings reported to
Who inspects GLP facilities? Who do they report to?
FDA
The Animal Care Facility should be of sufficient size to assure what 4 things?
separation of species
isolation of individual projects
quarantine of animals
routine or specialized housing of animals
In the Animal Care Facility, there should be separate space for experiments with test and control articles known to be ____. There should also be a separate area for ____ of laboratory animal diseases. There should also be facilties to handle ____.
biohazardous (volatile substances, aerosols, radioactive materials, infectious agents)
diagnosis, treatment, and control
disposal of animal waste
Storage areas, for feed (certified diets), nutrients, soils, bedding, supplies, and equipment,_____ and protected against infestation or contamination. Perishable supplies shall be ____ by appropriate means.
separated from areas where the test systems are located
preserved
Again, separate areas for what three phases of test articles? Storage of all test articles should be separate from ___.
Receipt/storage of test/control articles
Mix of test/control articles w/ carrier
Storage of test/control article mixtures
test systems
Should the lab space for procedures (routine or specialized)?
Yes
There should also be a separate space for the ___ that is limited to authorized personnel only.
archives
Equipment should be cleaned and maintained. If this equipment generates measurements, it should be ___, ___, and ____. Equipment should have written ___ and ___.
tested, calibrated, and standardized
SOPs and records (inspection, testing, findings, nature of defect - how discovered and how fixed)
Any GLP study needs SOPs - if there is any deviation from the SOP it should be authorized and documented by the ___. Significant changed need authorization written by ____.
Study director
management
What are things that need an SOP?
(1) Animal room preparation.
(2) Animal care.
(3) Receipt, identification, storage, handling, mixing, & method of sampling of the test & control articles.
(4) Test system [ie animals] observations.
(5) Laboratory tests.
(6) Handling of animals found moribund or dead during study.
(7) Necropsy of animals or postmortem examination of animals.
(8) Collection & identification of specimens.
(9) Histopathology.
(10) Data handling, storage, & retrieval.
(11) Maintenance & calibration of equipment.
(12) Transfer, proper placement, & identification of animals.
What can be used to supplement and SOP and how long must SOPs be on file?
-published literature
-historical file of all SOPs and revisions made should be maintained
Reagents and solutions should be labeled with what?
identity, titer, concentration, storage requirements, exp date
GLP - Newly received animals should be ____
isolated and health status evaluated
GLP - Study animals should be free of any disease. If they contract a disease they should be isolated and treated provided that ____. What should be documented and retained?
Treatment does not interfere with study
diagnosis, authorizations of treatment, description of treatment, and each date of treatment shall be documented and shall be retained.
What animals are required to receive appropriate identification? All information needed to specifically identify each animal within an housing unit shall appear where?
warm blooded animals excluding suckling rodents
on outside of housing unit
Can animals of different species be housed in the same room? Can animals of the same species, but different studies be housed in the same room??
No
Usually no especially if inadvertent mix up could affect outcome of either study
Does GLP list specifics on when animal cages, racks, and accessory equipment should be cleaned and sanitized?
No, just says regularly
What two things given to animals should be analyzed periodically? Documentation of the analysis should be maintained as raw data.
Feed and water
If any pest control materials are used their use shall be ____. It it could potentially interfere with the study it can’t be used.
documented
The identity, strength, purity and composition or other characteristics which will appropriately define the test or control article shall be determined for ____ and shall be documented.
each batch
The stability of each article shall be determined ___ or ___, according to SOP.
before the study initiation OR periodically for each batch
Each storage container for a test or control article shall be labeled by ____
name, chemical abstract number or code number, batch number, expiration date, if any and where appropriate, storage conditions necessary.
Studies more than ______ weeks duration will retain reserve samples from each batch of test/control articles. ___ of articles shall also be tested for uniformity and concentration.
4
mixtures
In GLP, how to handle SOP revisions? old versions?
A historical file of standard operating procedures, and all revisions thereof, including the dates of such revisions, shall be maintained.
What is the order of importance? GLP Regs, Protocol, SOPs
GLP Regs > Protocol > SOPs
Who signs amendment in GLP? who signs final report in GLP?
-Amendments must be signed and dated by SD
-Final report must be signed by SD and individual scientists or other professionals involved
Test and control article handling - Procedure for proper storage, contamination free distribution, proper ID throughout distribution, receipt of each batch (include ____)
date and quantity of batch distributed/returned
Mixed articles w/ carriers - If it’s mixed with carrier, what testing should be done? When does it expire?
1. determine uniformity & periodically concentration
2. determine stability either (A) BEFORE study initiation OR (B) according to SOPs providing periodic analysis of articles
Expiration date must be CLEAR, if multiple components have expiration date – EARLIEST date shown.
What should be included in the protocol?
(1) A descriptive title and statement of the purpose of the study.
(2) Identification of the test and control articles by name, chemical abstract number, or code number.
(3) The name of the sponsor and the name and address of the testing facility at which the study is being conducted.
(4) The number, body weight range, sex, source of supply, species, strain, substrain, and age of the test system.
(5) The procedure for identification of the test system.
(6) A description of the experimental design, including the methods for the control of bias.
(7) A description and/or identification of the diet used in the study.
(8) Each dosage level, expressed in milligrams per kilogram of body weight or other appropriate units.
(9) The type and frequency of tests, analyses, and measurements to be made.
(10) The records to be maintained.
(11) The date of approval of the protocol by the sponsor and the dated signature of the study director.
(12) A statement of the proposed statistical methods to be used.
Any changes to the protocol should be documented, signed by ___, ___, and maintained with ____.
study director, dated, maintained with the protocol
Recording of data generated during the conduct of study should be ____ and ____ by person entering data. Any change in entries shall be made so as ______, ____, and _____.
dated on the date of entry and signed or initialed by the person entering the data. Any change in entries shall be made so as not to obscure the original entry, shall indicate the reason for such change, and shall be dated and signed or identified at the time of the change. (same)
-dated and signed/initialed
-not to obscure original entry, indicate reason for change, dated and signed or identified at time of change
The final report shall include what 14 things?
(1) Name and address of the facility and the dates on which the study was initiated and completed.
(2) Objectives and procedures stated in the approved protocol, including changes.
(3) Statistical methods used.
(4) The test and control articles identification.
(5) Stability of the test and control articles.
(6) A description of the methods used.
(7) A description of the test system used. Where applicable, the final report shall include the number of animals used, sex, body weight range, source of supply, species, strain and substrain, age, and procedure used for identification.
(8) A description of the dosage, dosage regimen, route of administration, and duration.
(9) A description of all circumstances that may have affected the quality or integrity of the data.
(10) The name of the study director, the names of other personnel, involved in the study.
(11) A description of the transformations, calculations, or operations performed on the data, a summary and analysis of the data, and a statement of the conclusions drawn from the analysis.
(12) The signed and dated reports of each of the individual scientists or other professionals involved in the study.
(13) The locations where all specimens, raw data, and the final report is stored.
(14) The statement prepared and signed by the QAU.
(b) The final report shall be signed and dated by the study director.
Any corrections or additions to the final report should be amended by whom?
study director
All raw data, documentation, protocols, final reports, and specimens (Except ____) generated as result of a nonclincical laboratory study shall be retained.
-obtained from mutagenicity tests, wet specimens of blood, urine, feces, biological fluids
Since the results from GLP studies need to be kept indefinitely in the archives, what can a facility do?
contract with commercial archive holder to provide repository for data. Raw data/specimen can be retained elsewhere if specific reference to location included
Who is responsible for archives? What is a key feature of the archives?
an individual
able to retrieve information quickly
FDA-RETENTION OF RECORDS- how long are documentation, raw data and specimens retained in the archives?
Whichever of the following is shortest:
If study is not submitted... 2 years from the date the study is completed/discontinued/terminated
5 years following results submission to FDA
2 years following application approval by FDA
Doesn't apply to investigational new drug (IND) studies or investigational drug exemptions (IDEs)... keep these for 5 years following results submission
How long should slide records be held for? What about wet specimens?q
-same amount of time as data records
-as long as quality of preparation affords evaluation but not longer than data records
What happens to the records if the testing facility goes out of business? Who needs to be notified?
all data/documentation transferred to archives of SPONSOR
notify FDA in writing of transfer
What should happen to the data collected by a disqualified testing facility?
should still be submitted to FDA even if NCL study will not be considered in support of the application
When can a testing facility be disqualified?
-failed to comply with GLP regs
-noncompliance adversely affects validity of NCL study
-lesser regulatory action have not/will not be adequate to achieve compliance with GLP
How is a facility notified that they may be disqualified? What happens next?
Commissioner issues written notice
hearing for disqualification conducted
When does the commissioner evaluate records of disqualification? What are the 2 results of this evaluation?
-after hearing or time for requesting hearing expires w/o request made
-either does or does not make findings required for disqualification
-Notified testing facility with copy of order
What happens if a facility is disqualified?
-each application for research/marketing permit weather approved or not relying on NCL study from disqualified testing facility is examined
-FDA determines if study is acceptable regardless of facility disqualification
What happens to an NCL study apart of an application that was approved and found to be conducted at a disqualified facility BEFORE it was disqualified?
Decide if study was acceptable or unacceptable
-If acceptable regardless of facility disqualification - may require establish that study was not affected by circumstances that led to disqualification
-if study is unacceptable - data is eliminated from application - may justify termination/withdrawal of approval of application
What happens to an NCL study that began after date of facility disqualification?
study will not be considered in application, but does NOT mean you can’t submit the data gathered from the study
The Commissioner of the FDA can let other interested persons know of facility disqualification. What happens if the notice is sent to another federal government agency vs another person? Can the public see these records?
-Government agency - FDA will recommend agency consider whether or not NCL study should be accepted
-another person - FDA is not advising any action be taken by person notified
-yep
What are alternatives to disqualification?
-civil or criminal judicial proceedings or other regulatory action CAN HAPPEN AT SAME TIME
-FDA can refer another federal, state, or local government law enforcement or regulatory agency for action
-can refuse NCL study if study not conducted in accordance with GLP w/o disqualifying testing facility
If a sponsor terminates a testing facility from further participation in a nonclinical lab study as part of an application that has been submitted to the FDA, it shall notify the FDA in writing within ____ of the action
15 days
The Commissioner can approve reinstatement of a testing facility if they can prove compliance. To do this, the facility must present what and to whom? If the Commissioner reinstates the facility, how is this done? is this disclosable to the public
-in writing to Commissioner
-reasons it should be reinstated, detailed description of corrective actions taken or intends to take, assure acts or omissions which led to disqualification will not recur
-After inspection - Commissioner must notify in writing testing facility & all persons notified of disqualification
-yes
What does ENVIRONMENTAL PROTECTION AGENCY (EPA) do?
Regulates chemicals and monitors compliance with environmental laws
water and air compliance
Federal Insecticide, Fungicide, Rodenticide Act (FIFRA) - 40 CFR part 160
Toxic Substances Control Act (TSCA) - 40 CFR part 792

What is the difference between Federal Insecticide, Fungicide, Rodenticide Act (FIFRA) and Toxic Substances Control Act (TSCA)?
A major difference between EPA FIFRA and TSCA is the process used to review new Substances. While FIFRA calls for extensive test data as part of a complex registration process, TSCA requires only a 90-day advance notice to produce a new chemical.
EPA-RETENTION OF RECORDS- how long are documentation, raw data and specimens retained in the archives?
Whichever period is longest:
5 years following submission of results
2 years following termination, discontinuation, or completion
Cage cards, which contain animal identification numbers, are considered raw data and must be transferred to the archives during or at the close of study.
Answer: F, cage cards are NOT considered raw data.
Which Act passed by Congress is referenced as part of GLPs?
Federal Food, Drug and Cosmetics Act, as amended
A testing facility cannot be considered a Sponsor. T or F?
F- it can be if it initiates AND conducts the study
Testing facilities may be unexpectedly inspected by which governmental agency which will look specifically at records supporting an application for a research or marketing permit?
USDA
EPA
FDA
ISO
FDA
Deviations from SOPs are to be authorized by whom?
The Study Director
The Study Sponsor
The Testing Facility
The Quality Assurance Unit
The Study Director
Which animals do not need identification?
Adult mice
Guinea Pigs
Suckling Rodents
Farm animals
Suckling Rodents
Wet specimens such as blood, urine, feces, and biological fluids do not need to be maintained after the completion of a GLP study. T or F?
T
What types of studies are covered by Good Laboratory Practice regulations?
Clinical and Field Trials in animals
Non-Clinical Safety Testing
Basic Research Studies
Human Subject Trials
Non-Clinical Safety Testing
What is the law basis of DRUG ENFORCEMENT ADMINISTRATION (DEA)? Who administers it? Who enforces it?
Controlled Substances Act (CSA) - 1970 - Title 21 CFR 1300
Administered by the Secretary of Health and Human Services
Enforced by the DEA (est. 1973)
For which schedule drugs must supplier invoices be retained?
III-V
Schedule I-V drugs
- Schedule I: heroin, LSD, marijuana
- Schedule II: morphine, oxycodone, amphetamines, Oxymorphone, Pentobarbital(Nembutal), Fentanyl
- Schedule III: codeine mixtures (Tylenol #3), Telazol, Ketamine, Euthasol, Pentothal, Buprenorphine
- Schedule IV: diazepam, Darvon, Midazolam, tramadol, Phenobarbital
- Schedule V: codeine-containing cough syrup (pseudoephedrine)
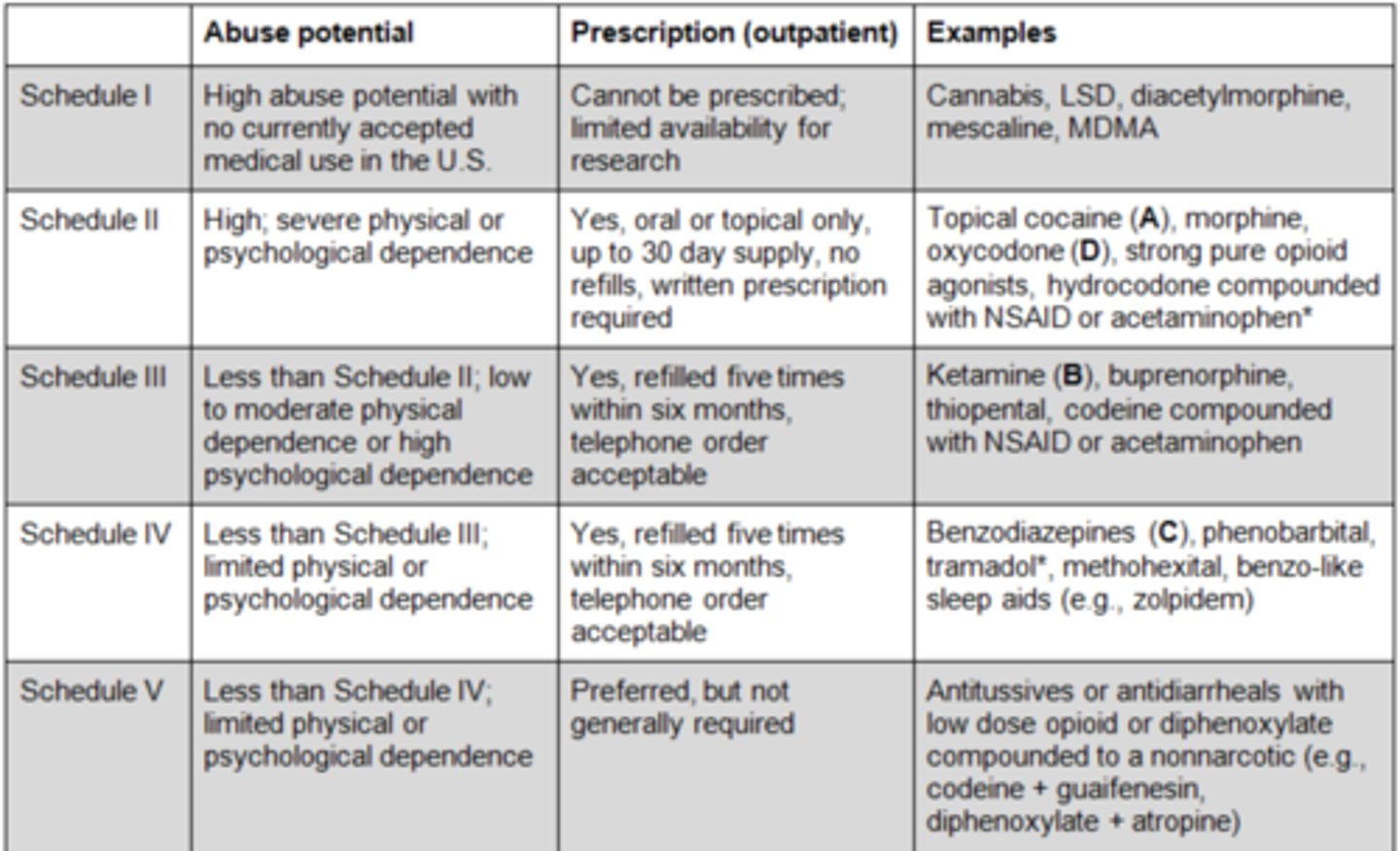
Schedule 1 drugs?
high potential for abuse, have no currently accepted medical use in treatment
heroin, LSD, marijuana, peyote
Schedule II drugs?
high potential for abuse, severe psychological or physical dependence
Cocaine
Codeine
Fentanyl
Hydrocodone
Methadone
Hydromorphone
Morphine
Opium
Oxymorphone
Pentobarbital
Sufentanil
Schedule III drugs?
moderate or low physical dependence or high psychological dependence
Buprenorphine
Hydrocodone (< 15 mg)
Ketamine
Opium combination product (<25 mg/du)
Thiopental
Tiletamine
Tiletamine + Zolazepam
Zolazepam
Schedule IV drugs?
limited physical dependence or psychological dependence
Butorphanol
Chloral Hydrate
Diazepam
Lorazepam
Midazolam
Phenobarbital
Alfaxalone
Tramadol