Molecular Impact of Variants
1/50
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
51 Terms
How can sequence variants affect GLP-1R function at the molecular level
Sequence variants (amino acid changes) can alter:
Ligand binding affinity (how strongly the agonist binds)
Receptor conformation (active vs inactive states)
Signal transduction (e.g., G protein coupling)
Molecular Dynamics Simulations
Molecular dynamics (MD) simulations are computational methods that model how molecular structures move and interact over time
atoms and molecules are allowed to interact for a period of time under known laws of physics
Simulate interactions between:
receptor (e.g., GLP-1R)
ligand (agonist)
Based on physical forces (bonding, electrostatics, etc.)
Molecular Dynamics Simulations: GLP-1R
MD simulations allow comparison of wild-type vs mutant receptors by analyzing:
ligand binding stability
conformational changes in the receptor
interaction networks (e.g., hydrogen bonds, contacts)
Interpretation:
Variants may weaken or strengthen binding interactions
Variants may alter receptor dynamics and activation states
Binding Affinity
describes how strongly a ligand (agonist) binds to a receptor
high affinity → strong binding
low affinity → weak binding
variants can disrupt key interactions (↓ affinity) or create new interactions (↑ affinity)
Dissociation Constant (KD) & Binding Affinity
KD is the ligand concentration at which half of the receptors are bound.
Low KD → high affinity (tight binding)
High KD→ low affinity (weak binding)
KD is an inverse measure of binding strength.
Relationship between KD and ΔG∘
∆Gº = RT ln(KD)
more negative ∆Gº → stonger binding (low Kd)
larger KD → less favorable binding
key idea: binding affinity is a thermodynamic property
relationship b/w Kd and ∆Gº is logarithmic: a small energy change (e.g. one hydrogen bond) leads to a 10-fold change in Kd
Implications:
Minor structural changes (e.g., side chains, H-bonds) can drastically alter binding
e.g. changes in amino acid side chains or in overall conformation can change the likelihood of two proteins binding to each other
Enables fine-tuning of specificity in biological systems

Binding Specificity
Binding specificity is the ability of a protein to prefer one ligand over others.
Determined by:
shape complementarity
chemical interactions (H-bonds, charges, hydrophobicity)
Variants can:
reduce specificity → off-target binding
increase specificity → more selective interaction
Binding Specificity Thermodynamics
specificity depends on the difference in binding free energies between ligands
∆GºB - ∆GºA
If binding to B has more negative ΔG∘ → B is preferred
Larger difference → higher specificity
Even small energy differences can strongly bias binding toward one ligand
Molecular force fields & minimization (MD basics)
Force fields (FF): mathematical models describing atomic interactions
include bond, angle, electrostatic, and van der Waals terms
Energy minimization:
adjusts structure to lowest energy conformation
removes steric clashes before simulation
Binding Equilibrium and Kd
at equilibrium, the rate of binding equals the rate of dissociation: Kon[A][B] = Koff [AB]
the dissociation constant is KD = Koff / Kon = ([A][B])/[AB])
stronger interactions shift equlibrium toward the complex (AB)
Fractional Occupancy
Fractional occupancy describes the fraction of receptor (A) bound to ligand (B):
fractional occupancy = [AB]/[A]total
[A] = [AB] when half of A is bound to B
using the equation from the previous slide, Kd = [B] under these conditions
initially occupancy increases significantly, then levels off
![<ul><li><p>Fractional occupancy describes the fraction of receptor (A) bound to ligand (B): </p><ul><li><p>fractional occupancy = [AB]/[A]<sub>total</sub> </p></li></ul></li><li><p>[A] = [AB] when half of A is bound to B</p></li><li><p>using the equation from the previous slide, K<sub>d</sub> = [B] under these conditions</p></li><li><p>initially occupancy increases significantly, then levels off</p></li></ul><p></p>](https://assets.knowt.com/user-attachments/d1318e03-37db-42ab-a8bd-3b090fbe718c.png)
Saturation and Binding Curves
as ligand [B] conc. increases, occupancy rises rapidly at first, then levels off (saturates) as receptors become fully bound
if [B] » Kd: system is saturated, nearly all receptors in bound state (AB)
if [B] = Kd: 50% occupancy
binding follows a saturation curve: fast increase → plateau when receptors are fully occupied
Binding Isotherm (fractional occupancy equation)
f = [L] / ([L] + Kd)
f = fraction of protein bound to ligand
[L] = ligand concentration
f = 0 → no binding
f = 1 → full saturation
f = 0.5 when [L] = Kd
This equation describes how binding increases with ligand concentration.
Binding Isotherm Curve
The plot of f vs [L] is a rectangular hyperbola:
Low [L]: very little binding (f≈0)
Intermediate [L]: rapid increase in binding
High [L]: saturation (f≈1)
Important point:
At f=0.5 → [L]=KD
Promiscuity
off target interactions results from low specificity
How do ideal binding affinity and KD depend on biological function? Case A
Permanent Complex
the affinities of the partners are likely to be high
low Kd
binding is near saturation → stable complex
importantly the dissociation constant is lower than the endogenous concentrations of the components so that binding will be close to saturation
Kd « cellular concentrations
How do ideal binding affinity and KD depend on biological function? Case B
Signaling Complex
affinities of partners are not so high
dissociation constant is roughly equal/slightly higher than the endogenous ligand concentration
Why are weaker (moderate) affinities important for signaling interactions?
allows small changes in either ligand conc. or other modulating factors to lead to big changes in the fraction of ligand bound
allows interaction to be regulated
weaker affinities allow interaction to be more dynamic
Kd of an interaction is diefined by its off rate (Koff) divided by its on rate (Kon)
kon is limited by rate of diffusion, koff is limited by affinity
higher koff → shorter binding time, enables rapid signal termination and regulation
Why are weaker (moderate) affinities important for signaling interactions? Continued
In signaling systems (e.g., hormone–receptor like GLP-1R):
If affinity is too high (very low KD):
receptors are nearly always saturated
small changes in ligand concentration produce little additional response
system loses dynamic range (no “sensitivity window”)
If affinity is moderate ( KD [ligand]):
receptor occupancy changes strongly with small ligand changes
system becomes highly responsive and regulatable
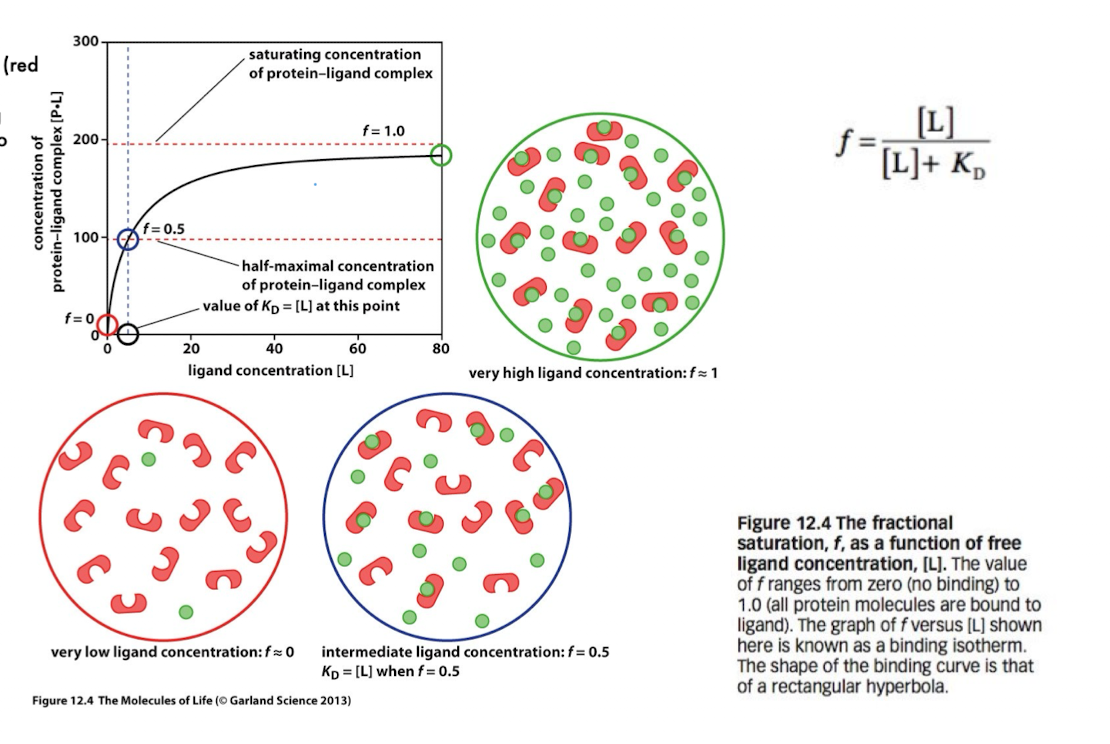
Competitive radioligand binding assay (how affinity is measured)
A fixed amount of radiolabeled ligand (e.g., 125I-exendin(9–39)) is bound to the receptor, then increasing concentrations of an unlabeled test ligand are added.
The test ligand competes with the radiolabeled ligand for receptor binding
As test ligand concentration increases → radioligand binding decreases
The concentration that reduces binding by 50% is the IC₅₀
Why are molecular dynamics simulations necessary instead of analytical solutions?
We cannot solve molecular behavior analytically because:
biological molecules have too many atoms and interactions
systems are too complex for closed-form equations
Instead,
MD uses numerical (step-by-step) comparison
calculates atomic motion over time, using physical force laws
What can we do w/ MD Simulations?
protein structure and dynamics: MD simulations help study protein folding, conformational changes and stability
we can use them to calculate free energies
enzyme mechanisms: MD cab help reveal atomic-level details of chemical reactions
membrane dynamics: simulations can be used to investigate behavior of lipid bilayers and membrane-proteins interactions
nucleic acids: they reveal the structure, flexibility, and interactions of DNA and RNA
drug discovery: they are used to model protein-ligand interactions, optimize drug candidates
What do we need for MD simulations?
an energy landscape (potential energy function) that describes how atoms interact
Defines forces between atoms (bonded + non-bonded interactions)
Includes effects of covalent bonds, electrostatics, van der Waals forces
Determines stable (low-energy) vs unstable (high-energy) configurations
ways to move the landscape
What does the molecular energy landscape represent physically?
Atoms have a preferred equilibrium position (minimum energy state)
Displacement from this position creates restoring forces
pulling atoms back together or pushing them apart
Bonds and molecular orbitals act like springs maintaining structure
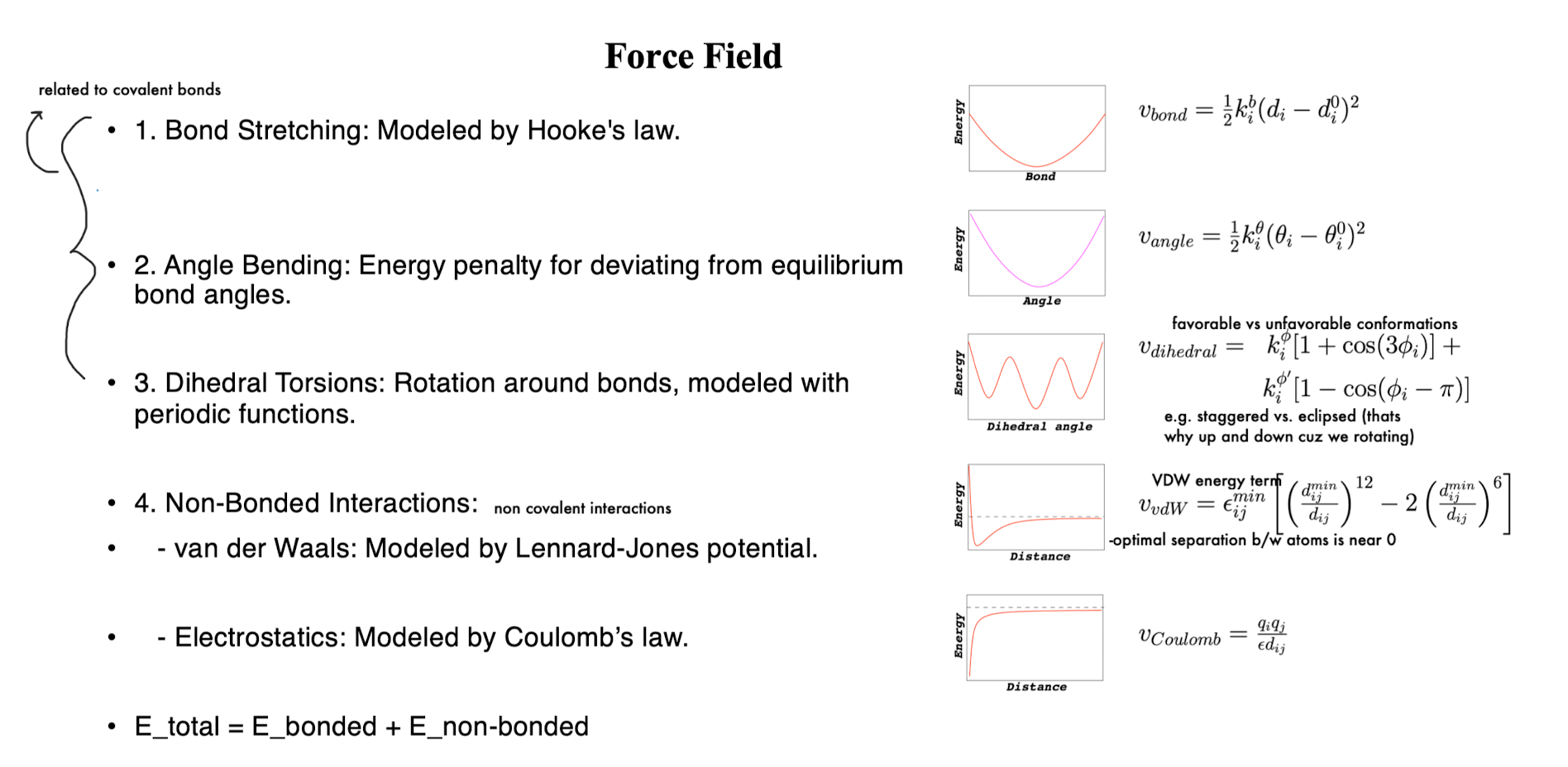
Force Field
a set of mathematical functions and parameters used to model the interactions b/w atoms in a molecular system
purpose: approximate the potential energy of a system based on atomic positions
types of forces:
covalent: bonded (bonds, angles, dihedrals)
non-covalent (VDW, electrostatics)
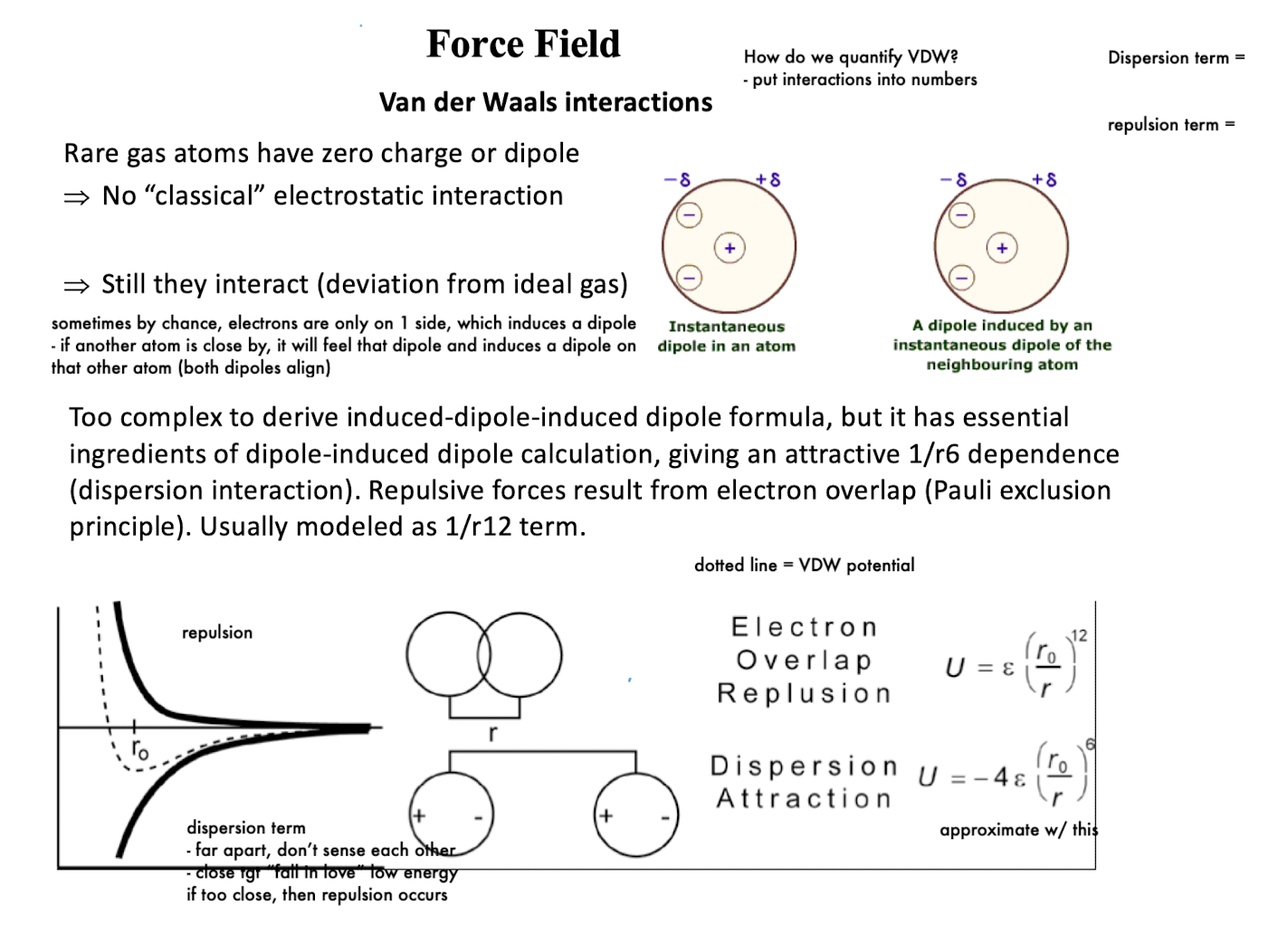
How are VDW interactions modeled in force fields?
dispersion term: 1/r6; repulsion term (electron overlap/Pauli exclusion): 1/r12
Far distance: no interaction
Intermediate distance: attraction (“optimal binding distance”)
Very close: strong repulsion (electron overlap)
VDW interactions balance attraction (dispersion) and repulsion (Pauli exclusion) to define stable atomic spacing
Force Field: Reference Point
the geometry where energy is lowest (most stable)
the further away, the higher energy penalty
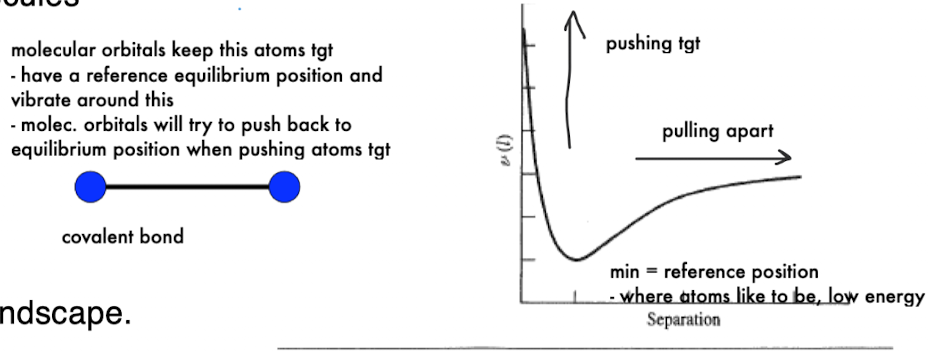
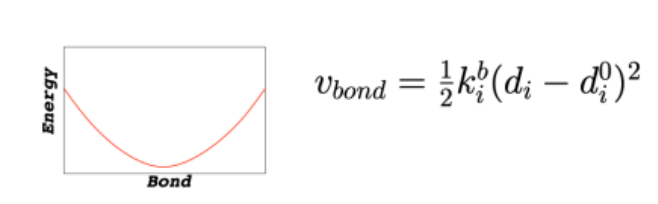
Force Field: Bond Stretching
modeled like a spring: E = ½ k(r-r0)2
r0 = ideal bond length (reference point)
energy increases symmetrically if bond is stretched or compressed
k = stiffness (steeper curve = harder to stretch)
narrow/steep curve → strong bond
wide curve → flexible bond
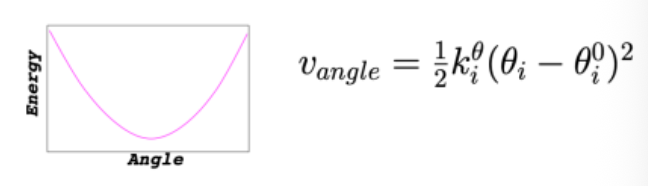
Force Field: Angle Bending
minimum at equilibrium angle; energy increases as you bend away from ideal geometry
Force Field: Dihedral Torsions
energy changes periodically as bond rotates
multiple minima = multiple stable
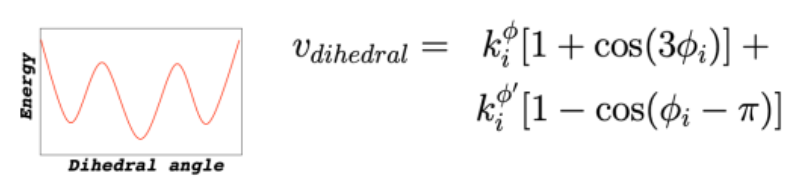
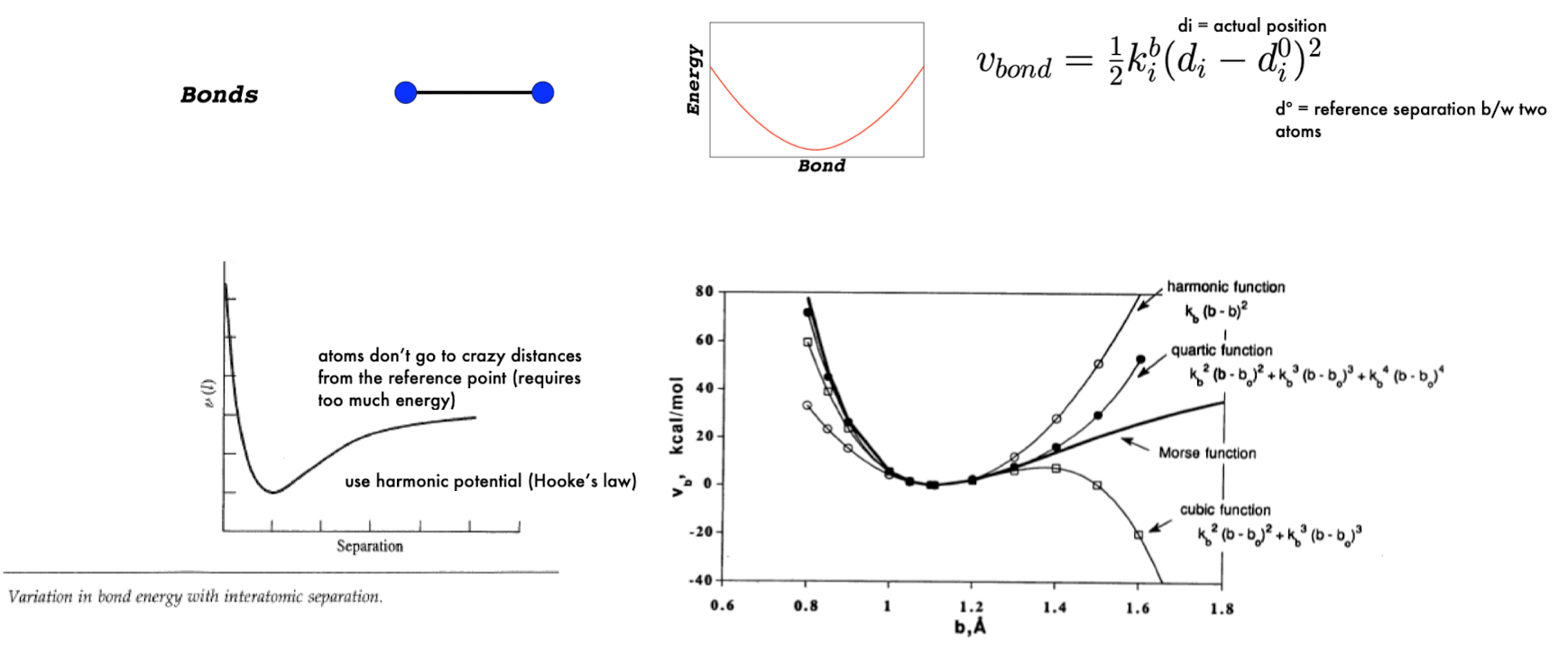
Force Field: Non-Bonded Interactions
VDW (Lennard-Jones potential)
V = potential energy
Interpret the curve:
far apart: V ≈ 0 (no interaction)
intermediate distance: negative energy (attraction)
too close → sharp increase (repulsion), very high V
sweet spot: minimum (most stable separation b/w atoms)
distance where attraction = minimum
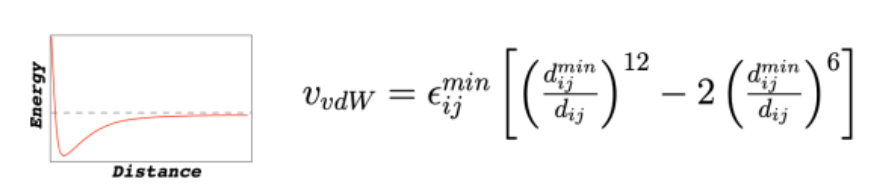
Force Field: Coulomb’s Law (Electrostatics)
opposite charges → negative energy (attraction)
same charges → positive energy (repulsion)
Stronger charges / closer distance → stronger interaction

Force Field Parameterization
process of deriving the parameters (bond lengths, angles, charges)
methods: fitting to experimental data (e.g. crystals structures, spectroscopy)
quantum mechanical calculation: how does energy change when pulling/pushing atoms tgt to get K
Challengs:
balancing accuracy and computational efficiency
Limitations of Force Fields
1. Approximations in the mathematical model: interactions are simplified mathematical forms
2. Difficulty modeling:
Polarizability.
Complex chemical reactions.
3. Dependence on quality of parameterization: accuracy depends on how well force-field parameters are fitted to experimental data
4. Computational cost for large systems
What is energy minimization in molecular dynamics?
Energy minimization is the process of finding a stable (low-energy) molecular structure on the energy landscape.
Atoms start in an initial configuration (often not optimal)
The system is adjusted to reduce total potential energy
Result = local energy minimum (stable conformation)
How is energy minimization in MD similar to linear regression?
Both are optimization problems:
Linear regression: find weights www that minimize loss (error)
MD energy minimization: find atomic positions that minimize potential energy
Analogy:
weights www ↔ atomic coordinates
loss function ↔ energy function
best fit line ↔ lowest-energy structure
Why can’t energy minimization in molecular systems be solved using standard calculus?
In molecular systems:
Energy depends on many variables (x, y, z for every atom)
There are many interacting terms simultaneously (bonds, electrostatics, VDW, etc.)
Improving one interaction can worsen another
Because of this:
you cannot simply take derivatives and solve analytically like in simple functions
the system is high-dimensional and highly coupled
How is energy minimization performed in molecular dynamics simulations?
Energy minimization is done using numerical optimization methods:
Start from an initial structure
Move atoms in small iterative steps
Always move “downhill” in energy
Limitations:
only finds a local minimum (closest low-energy state)
does NOT guarantee the global minimum (lowest possible energy)
different starting points can lead to different results
Gradient (Steepest) Descent
the steepest descent method minimizes a function by moving iteratively in the direction of the steepest negative gradient
key idea is to minimize a function by following the direction of maximum decrease
At each step, compute the gradient (slope) of the function
Move in the negative gradient direction (downhill)
Repeat until reaching a minimum
Steepest Descent Method: Advantages vs Limitation
Advantages: simple and intuitive, effective for small-scare minimizations
Limitations: convergence can be slow near minima
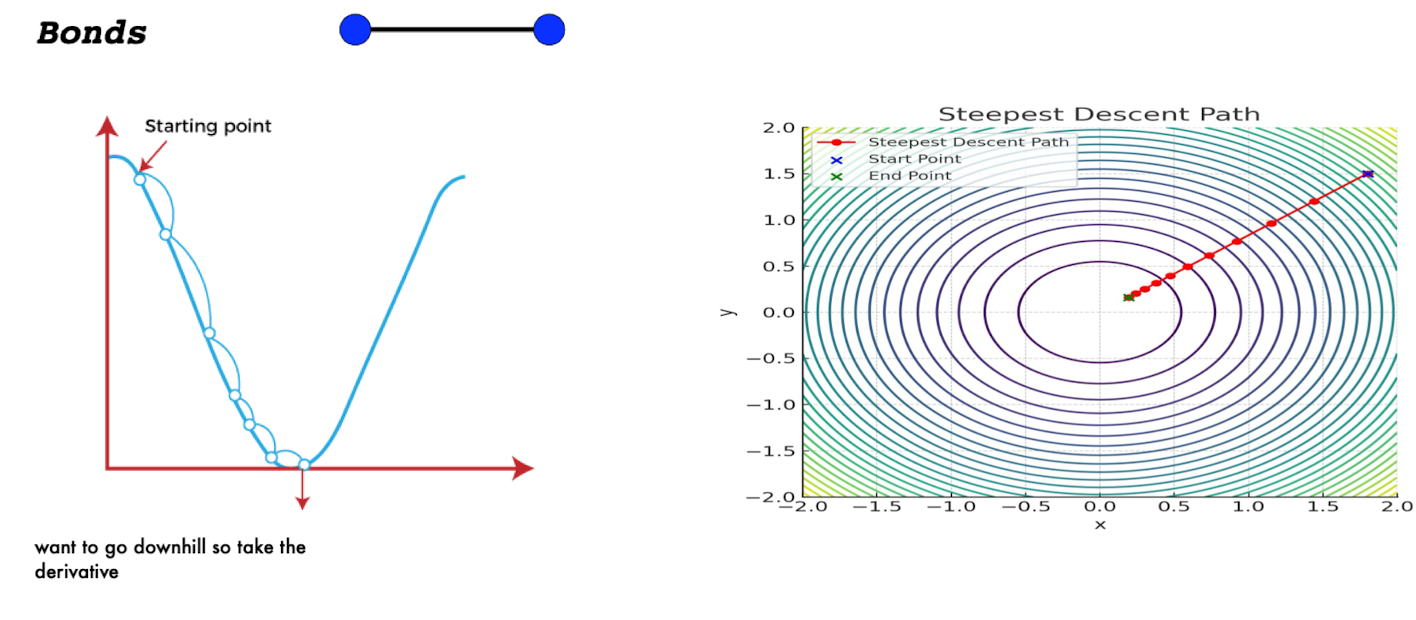
How steepest descent works?
compute gradient ∇(xk) at the current point rk
choose direction: move in the direction -∇(xk)
choose step size: ⍺k determines how far to move
update position: Xk+1 = xk - ⍺∇f(xk)

How does steepest descent relate to energy minimization in molecular systems?
Start with a molecular structure
Use a force field to compute energy
Compute the gradient of energy → gives direction of force
Move atoms in direction that lowers energy