(L6) IMED2002 - Anaemia - Abnormal RBC Production
1/40
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
41 Terms
Haemolytic anaemia can lead to destruction of red
cells in the blood vessel or outside the blood.
Which of the following is an indicator of
intravascular haemolysis?
A. Anaemia B. Elevated bilirubin C. Haemoglobinuria D. Reticulocytosis
C. Haemoglobinuria
Haemoglobinuria (C): When RBCs break down inside the vessels, free haemoglobin is released. When this exceeds the binding capacity of the protein haptoglobin, the free haemoglobin is filtered by the kidneys and excreted into the urine, turning it dark-brown or red. This is a classic, direct indicator of intravascular haemolysis.
.
Anaemia (A), Reticulocytosis (D): These occur in both intravascular and extravascular haemolysis as the bone marrow tries to compensate for the loss of cells.
Elevated bilirubin (B): This is a marker of haemolysis in general, but is typically more pronounced in extravascular haemolysis (destruction in the spleen/liver
1. Normal Haemoglobin Structure
- Normal adult Hb consists of two alpha and two non-alpha global chains
- HbA 98% (alpha 2 beta 2)
- HbA(subscript)2 is around 2% (alpha 2 delta 2)
- HbF <1% (alpha 2 gamma 2) (fetal)
.
- Fetus: Hb synthesis begins with zeta and epsilon global chains
- By birth: majority is HbF (switch from HbF to HbA takes place by 6 months)
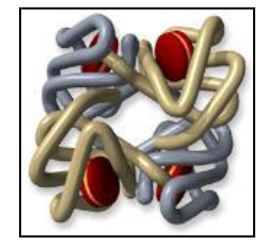
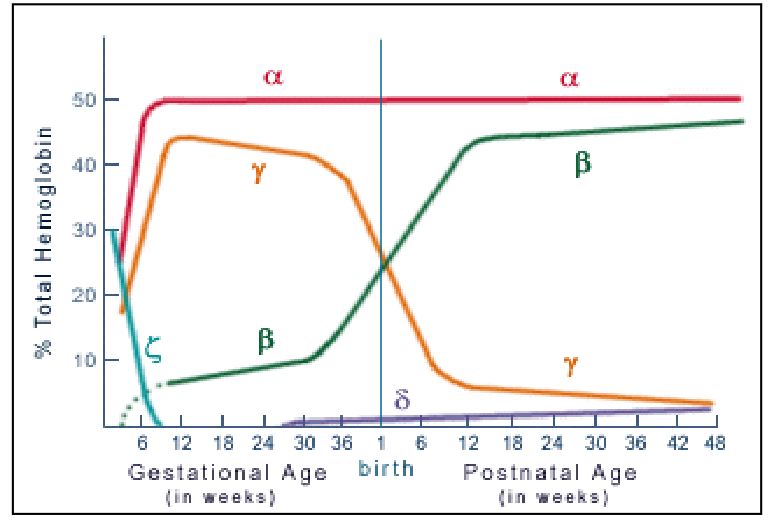
Embryonic, Fetal, Neonatal and Adult Haemoglobins
DIAGRAM ON SLIDE 5
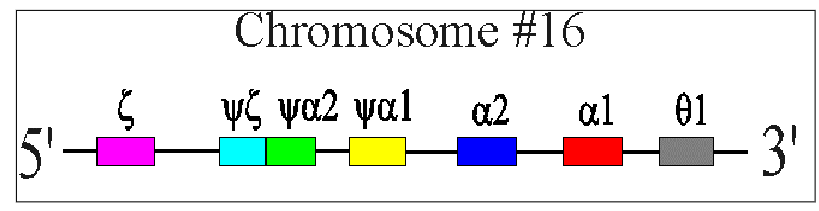
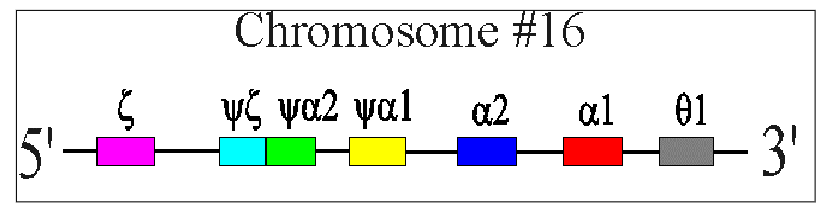
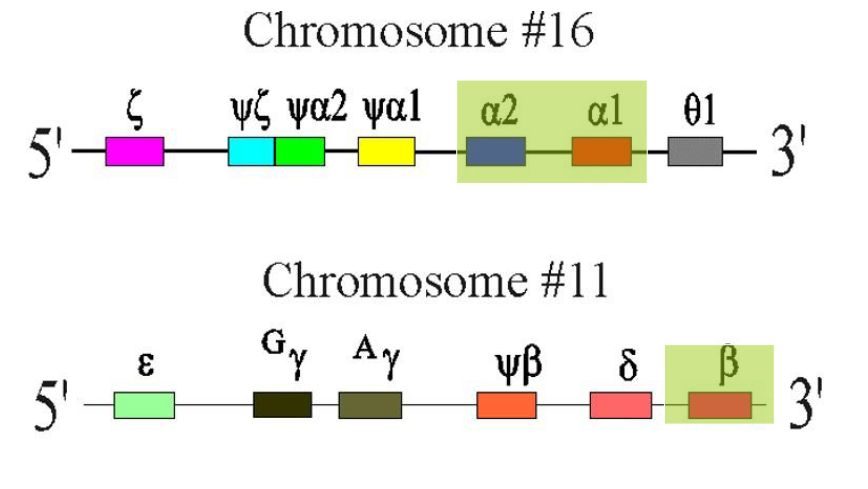
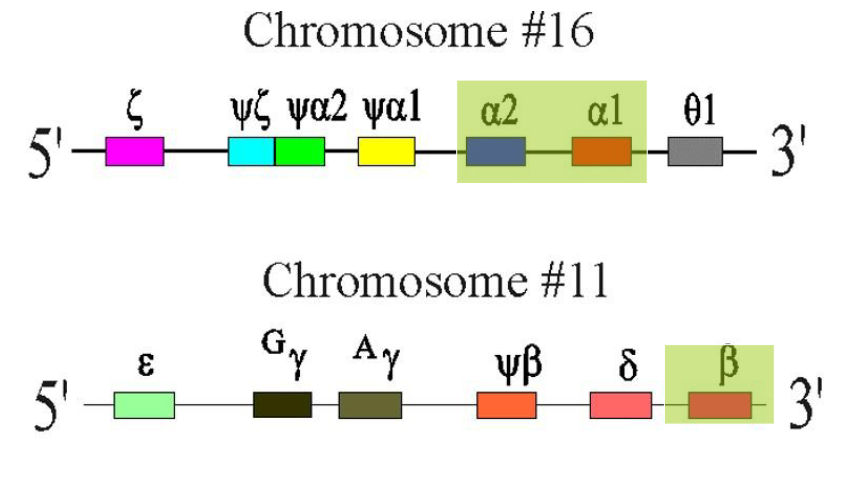
alpha-Globin Chain Synthesis
ALPHA GLOBIN CHAINS:
- Two alpha-globin genes on each chromosome 16 (total of 4 genes therefore)
- Each alpha globin gene is responsible for 25% of the normal total alpha-globin chain synthesis
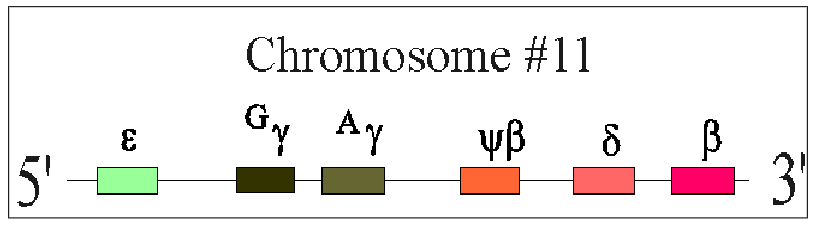
beta-Globin Chain Synthesis
NON-ALPHA GLOBIN CHAINS:
- One Beta-globin gene on each chromosome 11 (total of 2 genes total)
- Beta gene is part of a complex containing delta and gamma genes
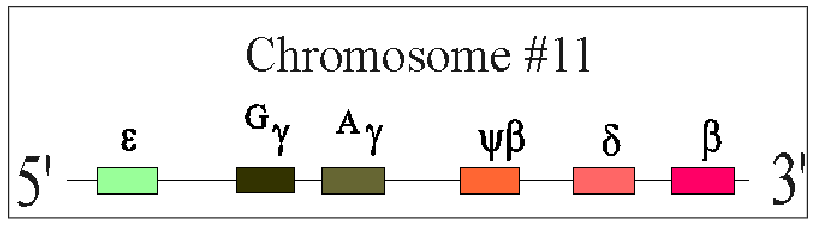
2. Haemoglobinopathies
- Genetic disorders of Hb synthesis
- Abnormalities of alpha or beta-globin chain synthesis
- Microcytic red cells and reduced RBC survival
.
THALASSAEMIA:
- Reduced production of alpha or beta globin chains
- alpha or beta thalassaemia
.
STRUCTURAL HAEMOGLOBINOPATHIES:
- Abnormal globin chain structure
- HbS (sickle cell disease)
Thalassaemia
- Commonest monogenic disorder in the world
- High frequency in Mediterranean, Middle East, Asia
- Reduced synthesis of one or more globin chains
- Imbalance between amount of alpha and beta globin chians
- RBC damage from efects of globin chain in excess
.
- esentially its a reduced synthesis of one or more of the globin chains that results in imbalance between the amount of alpha and beta
- becuase of that imbalance, the red cells are not being produced or not making normal haemoglobin
.
- blood film shows that the person with thalassaemia has red cells that are paler and smaller then normal and sometimes can be dificult to distinguish between this and iron deficiency
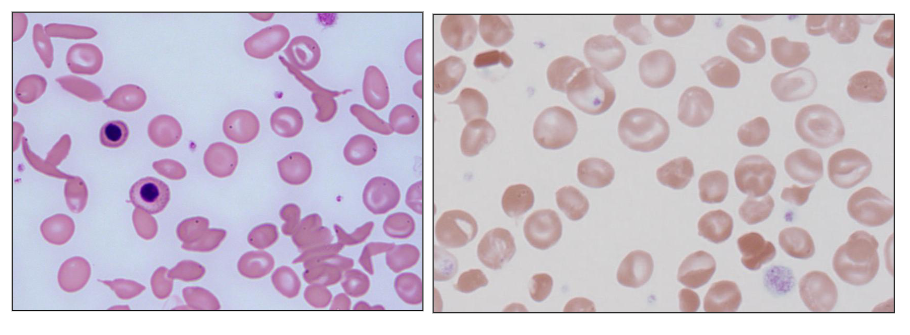
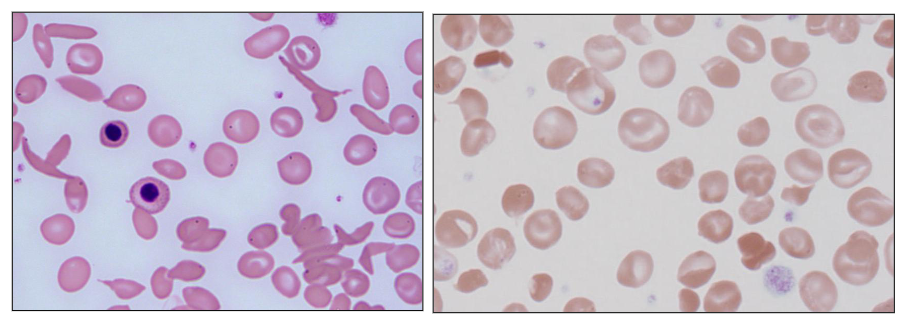
Structural Haemoglobinopathies
- Point mutation resulting in amino acid substitution
- Structurally abnormal Hb (haemoglobin)
- Clinical features dictated by properties of the abnormal Hb from the amino acid substitution
- E.g sickle haemoglobin (HbS); target cells: HbE, HbC
- on the left, the red cells are shaped like a sickle

PUT THE IMAGE ON SLIDE 12 AS A QUESTION
- answer is C


Thalassaemia Background
- Thomas Cooley described Thalasasmia in 1925
- Italian children with severe anaemia, enlarged spleen and distinctive appearance due to bone deformities (facial)
- High prevalence around Mediterranean sea
- deficienct synthesis of globin chains of Hb
- Classified by clinical severity

Geographic Distribution
- Commonest genetic disorder worldwide
- 3% world population affected by beta thalassaemia (Mediterranean basin; SE Asia; W Africa as well as Thailand 4.8-10% population (alpha-thalassaemia)
- prevalent in iltay and greece
.
- migration patterns: prsence in australia
Pathophysiology
- reduced amount of alpha or beta globin chain
- red blood cells are abnormal; reduced survival
- increased bone marrow erythropoietic response
- Severity: mild - moderate - severe - fatal
.
ALPHA-THALASSAEMIA: gene deletions
- Common: 3.7kb; 4.2kb; -SEA deletions
- reduced alpha globin chain production
.
BETA-THALASSAEMIA: point mutations
- Numberous mutations: different clinical severity
- Reduced Beta-globin chain production
Clinical Features
Severe: incompatible with life (Barts hydrops)
Anaemia: variable severity from
- Transfusion dependent (beta-thalassaemia major) to mild or asymptomatic (thalassaemia minor)
.
Severe forms:
- Stunted growth, endocrine, iron overload
- Mild splenomegaly, hepatomegaly
- Infections, folate deficiency
- Stunted growth, bony deformities, fractures
- Iron overload: cardiac problems; endocrine failure
Which of the following is most correct in relation to thalassamia?
A. Due to an abnormality of haemB. It is incompatible with life C. Is a type of haemolytic anaemia D. Is always due to point mutations E. Is always due to gene deletions
C. Is a type of haemolytic anaemia
.
Thalassemias are a group of inherited disorders (hemoglobinopathies) characterized by reduced production of hemoglobin chains (α) or β), leading to microcytic, hemolytic anemia. The ineffective erythropoiesis (production) and premature destruction of red blood cells (hemolysis) define it as a hemolytic anemia.
PUT THE IMAGES ON DIAGRAM 18
DIAGRAM ON SLIDE 18

Beta-Thalassaemia: terminology
- decreased beta-chain production: beta+ thalassemia
- Absent Beta-chain production: B0 (beta nought) thalassaemia
- One gene abnormality: Beta thalassaemia minor (generally beta Beta +)
TWO GENE ABNORMALITIES: beta thalassaemia major
- B0 B0: most severe
- B0 B+: moderately severe (intermedia)
- B0 B: moderately severe (intermedia)
- B+ B+ : increased HbF and A2

Beta-Thalassaemia Major
- Dx: 3-6 months (when HbF no longer made and unable to make HbA (no beta chains)
- Symptomatic severe anaemia (Hb, 50g/L)
- extramedullary erythropoiesis: spleen
- Bone marrow expansion
TRANSFUSION DEPENDENT (lifelong):
- Iron overload
- Iron chelation therapy
- Long survivors; secondary problems (e.g bone thinning - osteopenia)
.
- Bone marrow transplantation
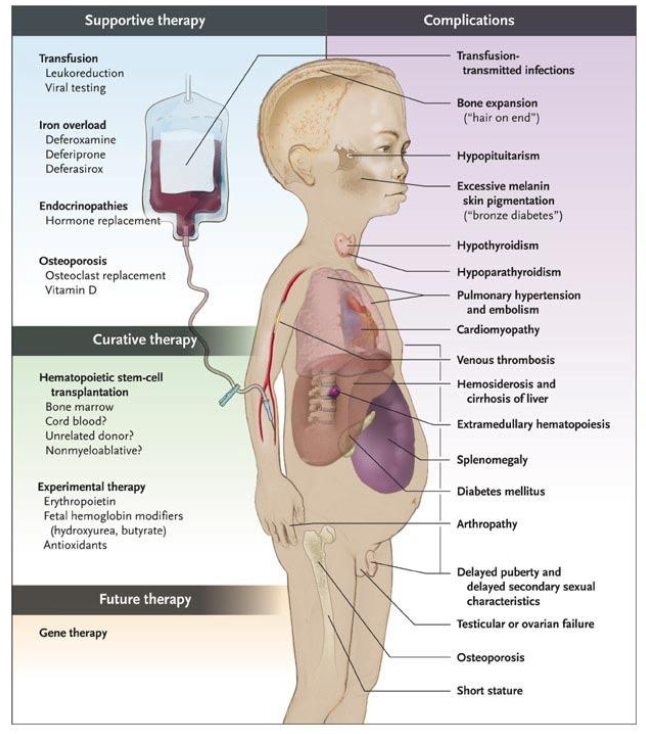
Thalassaemia Diagram
DIAGRAM ON SLIDE 23
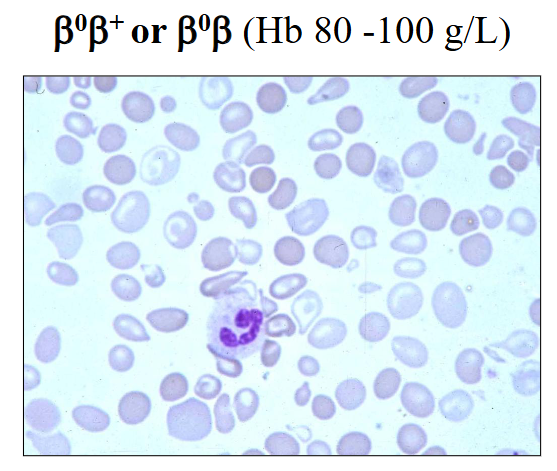
Beta-Thalassaemia Intermedia
- B0B+ or B0B (Hb 80 - 100g/L)
- symptomatic anaemia when 'stressed' (e.g sepsis; hypoxia)
- rarely require transfusions or splenectomy
.
- rarely require spleen to be removed (splenectomy) whereas patients with thalassaemia major may require a splenectomy
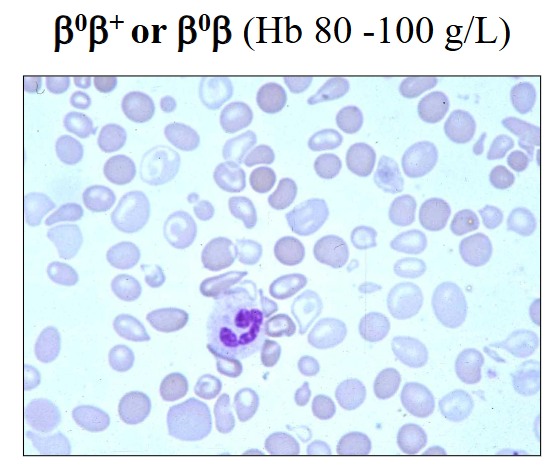
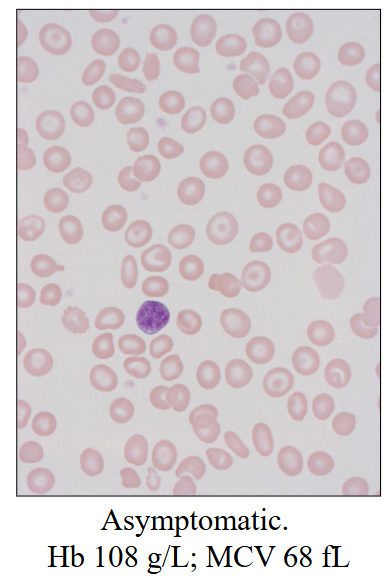
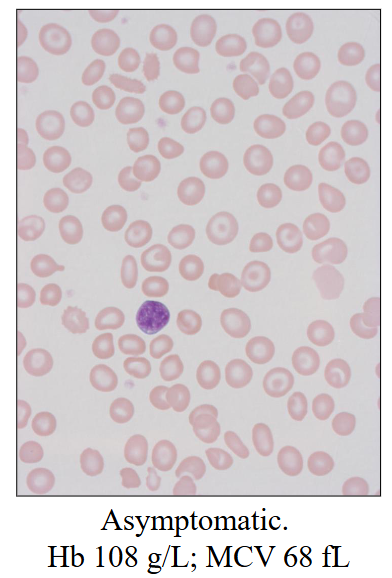
Beta-Thalassaemia Trait (minor)
- Usually B B+ - thalassaemia
- Borderline anaemia
- Microcytic red cells
- usually asymptomatic
- PB film: unremarkable or mild poikilocytosis
- Elevated HbA2
- Borderline elevated HbF

ADD QUESTION ON SLIDE 26
- answer is E

Why do patients with beta-thalassaemia major present at 6 months of age? Justidy your answer
- its because of the switching off of the gamma globin gene which commences prior to birth, but it only switches off by 3-6 months of age
- the beta globin gene, which should have switched on around 6 months of gestation and should be the main second globin chain that combines with alpha by 6 months of age cannot be produced
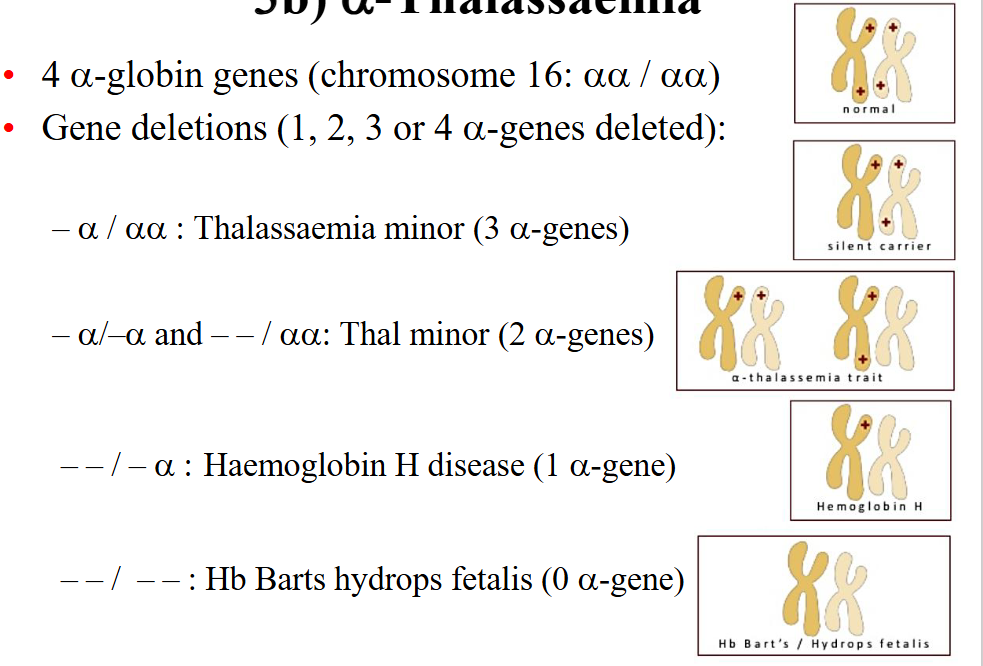
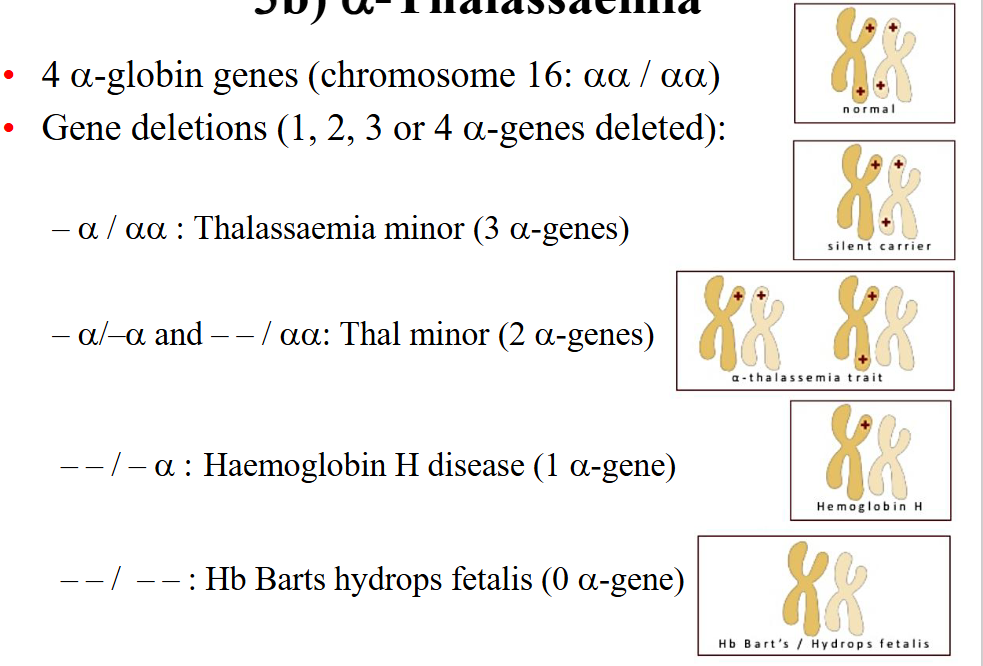
alpha-thalassaemia
- 4 alpha globin genes (chromosome 16: alpha alpha / alpha alpha)
- Gene deletions (1, 2, 3, or 4 alpha-genes deleted):
.
- a / aa: Thalassaemia minor (3 alpha genes)
- a / -a and --/aa : Thal minor (2 alpha genes)
--/-a: Haemoglobin H disease (1 alpha-gene)
--/-- : Hb Barts hydrops fetalis (0 alpha gene)
Alpha-Thalassaemia is due to ___ and beta thalassaemia is due to ____
deletions, point mutations
Severe: Hb Bart's Hydrops Fetalis
- First described in 1960; most severe form
- No alpha-globin genes on Chr. 16 (all alpha-genes deleted)
- No alpha-globin chains can be produced
- Usually fatal in utero (or premature delivery 30-34 weeks)
- Hydropic infant (severe swelling): due to cardiac failure from severe anaemia
- Blood: anaemia; erythroblasts
- Both parents: --/aa (thalassaemia minor); carriers (alpha to the power of 0)

HbH Disease
Only one α-globin gene: - - / - α (a^0/a+)
3 α-globin genes lost from deletions
SE Asia (--SEA), Mediterranean (--MED), Fil (--FIL)
Clinically: thalassaemia intermedia:
Moderately severe: Hb 60 - 100 g/L
Symptomatic when “stressed”
Chronic haemolytic anaemia: erythroid hyperplasia
Splenomegaly; cholelithiasis; leg ulcers
Splenectomy
Pregnancy: can have symptomatic anaemia

alpha-Thalassaemia Trait
Deletion of 1 or 2 α-globin genes
- -/αα or - α/- α (α0-thalassaemia)
- α/αα (α+-thalassaemia)
3.7 or 4.2 kb deletions most common
Haematology:
- Hb: Normal or mild anaemia
- MCV: 70-78 fL ; MCH <25 pg
- Film: normal / mild poikilocytosis

What is the most correct in relation to HbH Disease
D. Due to inheritance of abnormal chromosome 16 from both parents

Structural Haemoglobinopathies
- Point mutations causing structural abnormality in one
of the globin chains of Hb
Most common types we see:
HbS: African or Middle Eastern heritage
HbE: Thailand; target cells; low MCV; common
HbC: West Africa; target cells
Can occur together with a thalassaemia (“compound
heterozygote”) or another structural defect; e.g…
- HbE/beta-thalassaemia (can be severe)
- HbS/beta-thalassaemia
- HbSC
4a) Haemoglobin S
- Point mutation at codon 6 in β-globin gene (βS)
- Causes insolubility of HbS when deoxygenated
- Insoluble globin chains crystallise in red cells
causing the cells to “sickle”
- Ethnic background: Indian subcontinent, Middle
East, Equatorial Africa
- Most common in West Africa
- Homozygous: Sickle cell disease (HbSS)
- Heterozygous: Sickle cell trait (HbAS)
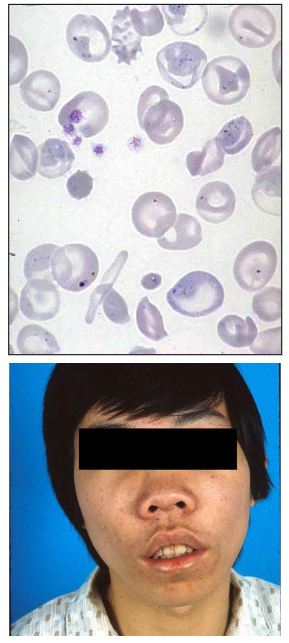
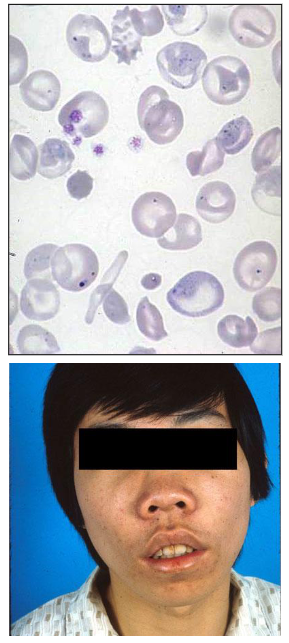
Sickle Cell Disease: HbSS
Homozygote (Hb SS):
- Haemolytic anaemia and sickling crises
.
- Vaso-occlusive events: due to sickling (blocked vessels)
• Pain, tissue infarction, leg ulcers, bone pain, stroke
• Acute chest syndrome, congestive heart failure
• Aplastic crises: marrow failure; anaemia
.
- Reduced splenic function: infections, meningitis,
Lowered resistance to salmonella and pneumococcus
- Gallstones
- HbS can be inherited with beta-thalassaemia (HbS/beta-thal)
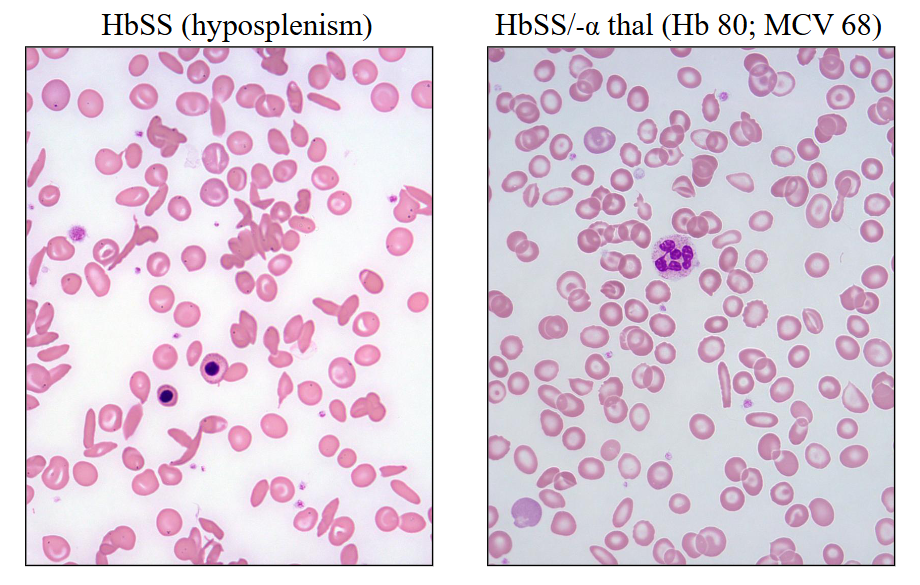
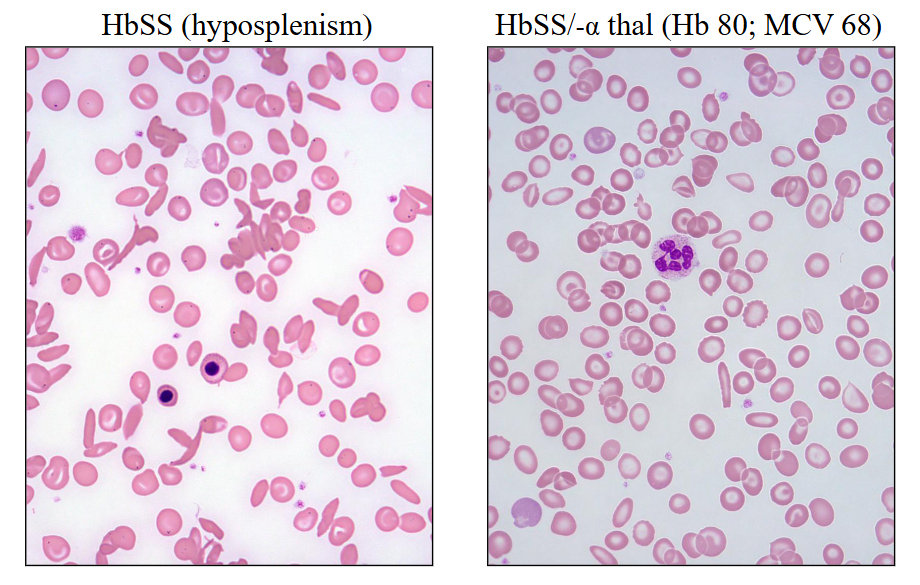
Sickle Cell Disease
HbSS (hyposplenism)
HbSS/-alpha thal (Hb 80; MCV 68)
Sickle Cell Disease
Laboratory features:
• Anaemia: Hb 70 – 90g/L
• Blood film: sickle-shaped red cells
.
Treatment:
• Avoid dehydration, infections
• Vaccination for pneumococcus and haemophilus
• Oral penicillin (indefinite)
• Folic acid: to prevent folate deficiency
• Transfusions: for severe anaemia
• Oral hydroxycarbamide: increases HbF (increases foetal Hb)
• Stem cell transplant
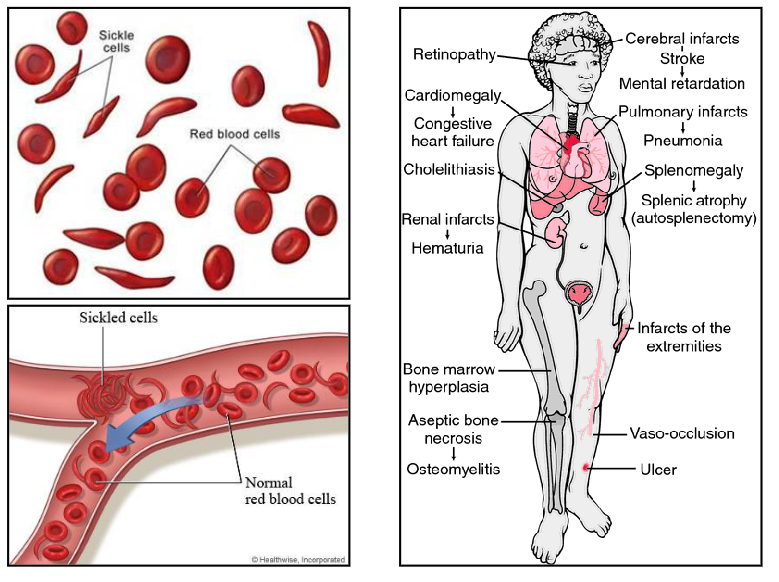
Sickle Cell Disease Diagram
DIAGRAM ON SLIDE 39
.
Sickle Cell Trait
- HbAS (carriers; heterozygous for HbS)
- Benign condition
- Normal Hb (no anaemia)
- Genetic counselling
- Natural resistance to malaria
- Sickle cell trait occurs at a higher frequency in
affected populations in the tropics
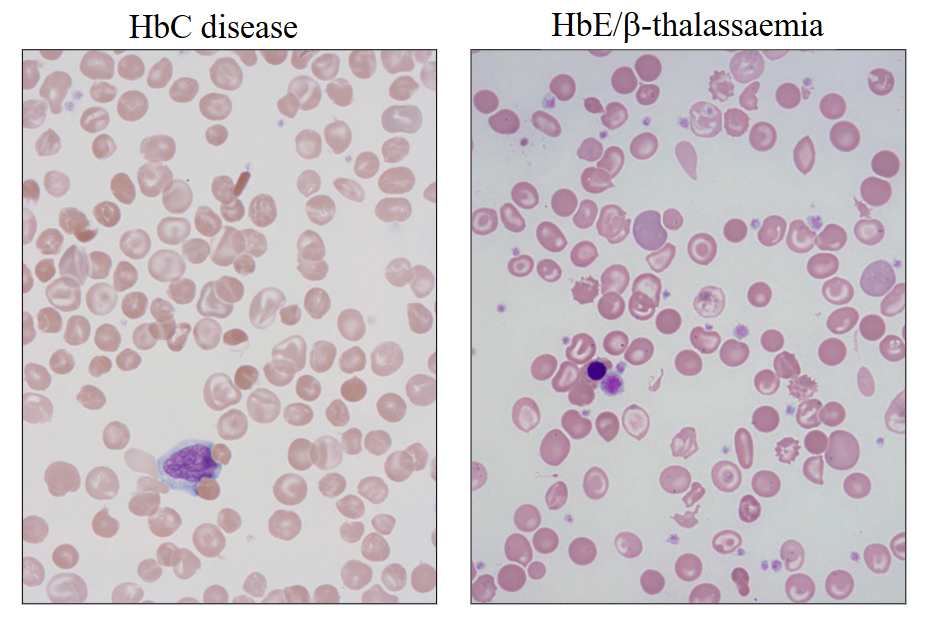
Other Haemoglobinopathies
HbC disease
HbE/beta-thalassaemia
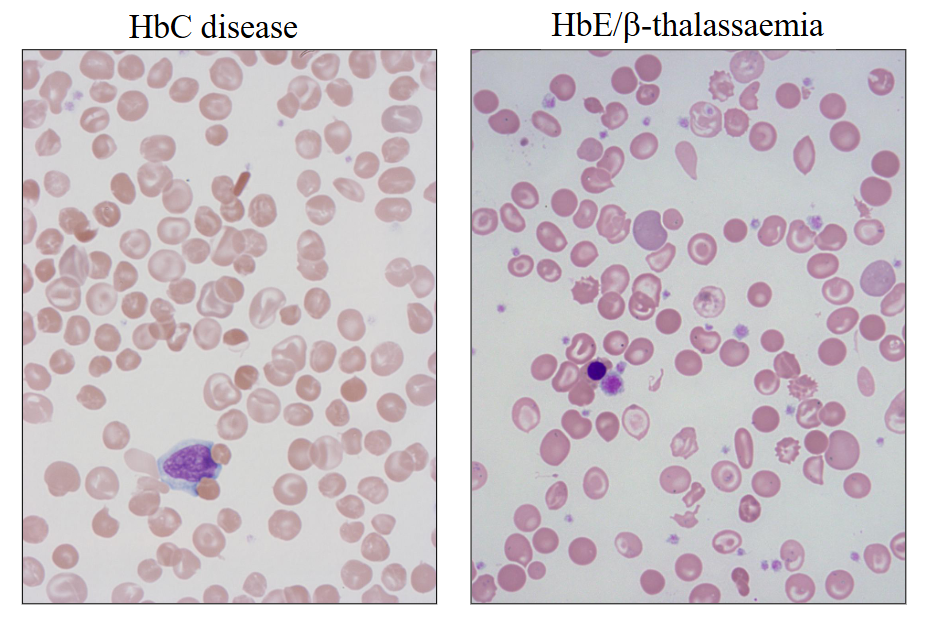
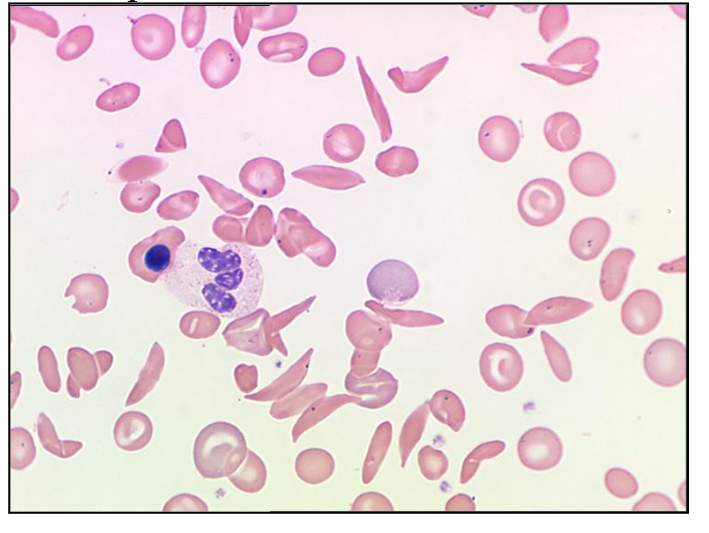
How many sickle cells are there in this picture of the blood film?
LOOK AT DIAGRAM
there are 20 (about 20, not exactly)
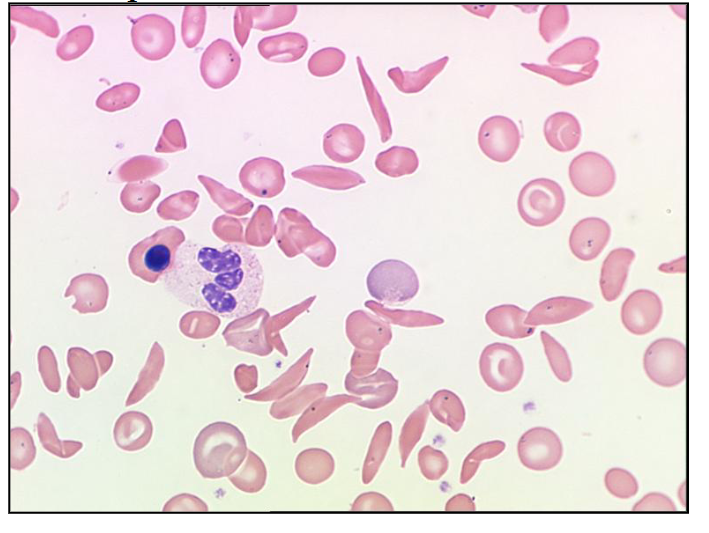
Sickle cell disease can cause
all of these features can occur
Acquired Disorders
- Abnormal bone marrow (incl. red cell) production
.
Primary:
- Myelodysplasia: bone marrow dysfunction (-dysplasia):
• Acquired genetic defect: mutation or chromosome
• Stem cell defect; anaemia or pancytopenia
• Older population
• Bone marrow shows abnormal cell morphology
• Can progress to leukaemia
.
Secondary:
- Drugs affecting bone marrow production: examples:
methotrexate; cytotoxics; antibodies
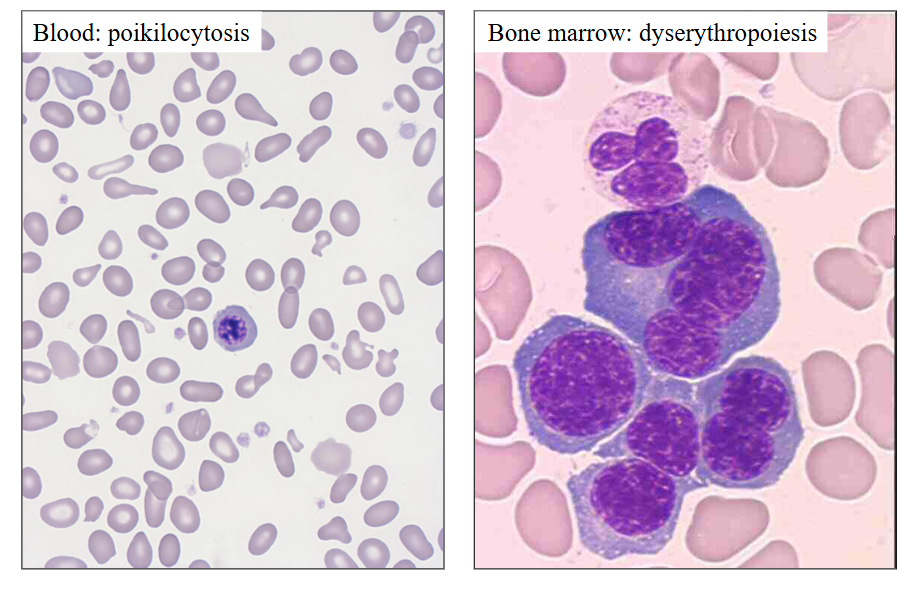
Dyserythropoiesis: myelodysplasia
DIAGRAM
.
myelo = marrow
dys = abnormal
plasia = making
abnormal blood cell production

Learning Outcomes
TEXT
