PCEU 509 Exam #2 (Review)
1/166
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
167 Terms
introduction to the epithelium
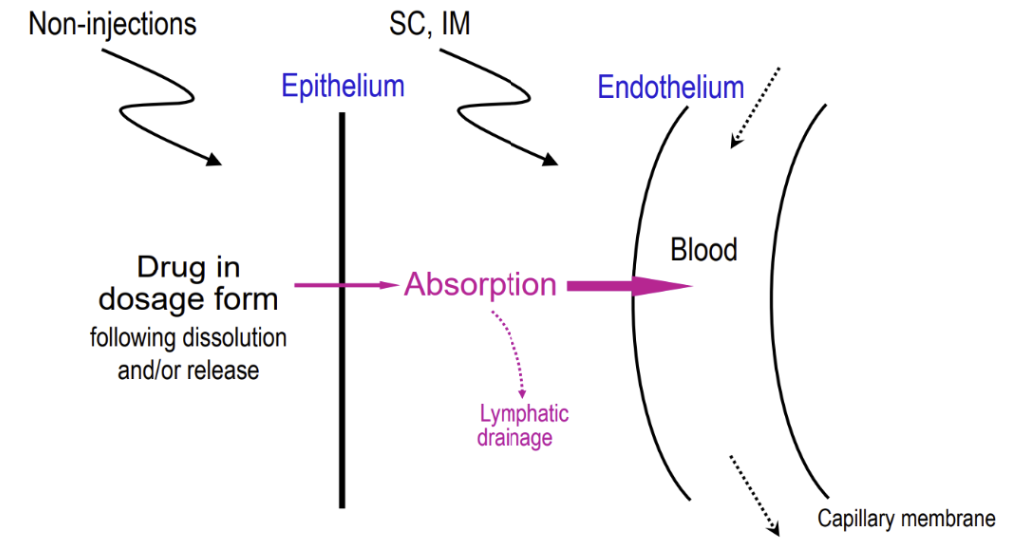
All internal and external body surfaces are lined with epithelium
To be absorbed, drugs must penetrate that epithelium to reach systemic circulation
Injections (IM, SC) are an exception because they can bypass the skin
After the epithelium, there is also fat, vasculature, etc. that needs to be passed to get to blood
For drug absorption in non-injection administration, what is the rate-limiting barrier?
epithelium
It can define whether a drug is or doesn’t get absorbed
This makes injection dosage forms the most effective way to administer the drug, but it’s not favored by many patients
types of epithelium
Simple squamous
Simple columnar/cuboidal
Stratified squamous
Pseudostratified columnar
Transitional
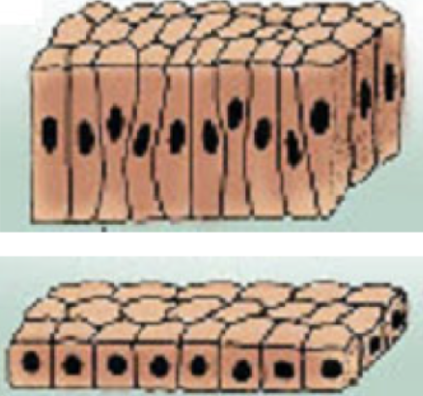
simple squamous epithelium
a single layer of thin flattened cells
Allows for good drug absorption
Found in: Lung’s alveoli and kidney’s filtration tubule
Endothelium (inner surface of blood vessels) is also similarly structured, which is what makes injections effective

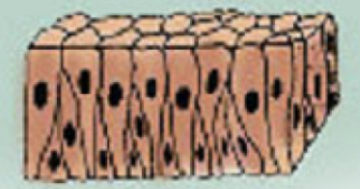
simple columnar/cuboidal epithelium
a single layer of column/cube-shaped cells; ciliated or nonciliated
Thicker than simple squamous, which makes it less good for drug absorption, but absorption still occurs since it’s not that much thicker
Found in: Lung’s bronchioles, kidney’s distal tubule and collecting duct, stomach, and small and large intestines
stratified squamous epithelium
multi-layers of flattened cells, with outer cells keratinized or non-keratinized
Much thicker than simple squamous and simple columnar/cuboidal
Found in: Oropharynx cavity, esophagus, rectum, vagina, cornea, and skin (keratinized)
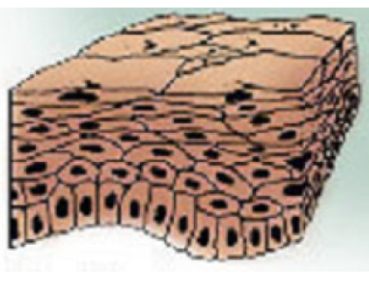
pseudostratified columnar epithelium
a single layer of columnar cells, each touching the basal lamina directly; their nucleus location gives a stratified look
Thickness is similar to simple columnar
Found in: Nose, larynx, trachea, and urethra
transitional epithelium
multilayers of differently shaped cells that allow stretching
Found in: Urothelium of the bladder and ureter
Rank the permeability of the different types of epithelium from most permeable to least.
Simple squamous > Simple columnar/cuboidal and Pseudostratified columnar > Transitional epithelium > Stratified squamous
components of epithelium that influence their permeability
Epithelial types vary across different body surfaces → Influence the kinetics of drug transport at each site
Cell thickness:
Simple < Stratified
Squamous < Columnar/cuboidal
Tightness between cell junctions
When cells are super squished together, that makes it harder to permeate
Functionality of specialized transport
cell membrane
Made up of two layers of phospholipids
A phospholipid itself has a hydrophilic head that faces toward extracellular and intracellular aqueous situations while the lipophilic tails face toward each other
Lipophilic molecules get taken in, while hydrophilic molecules are repelled
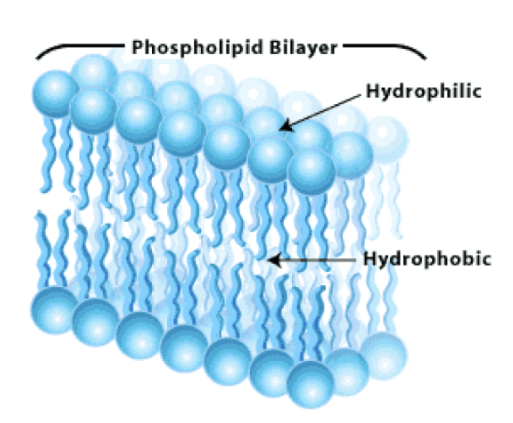
diffusive sorting by the cell membrane
We want drugs to be more lipophilic because they can get through the phospholipid bilayer without help
Despite water being hydrophilic, it’s just so small and also not charged, allowing it to still can get in
Ions are charged, hydrophilic molecules that can’t get through the phospholipid bilayer, so it needs a transporter/channel if it wants to get into the cell
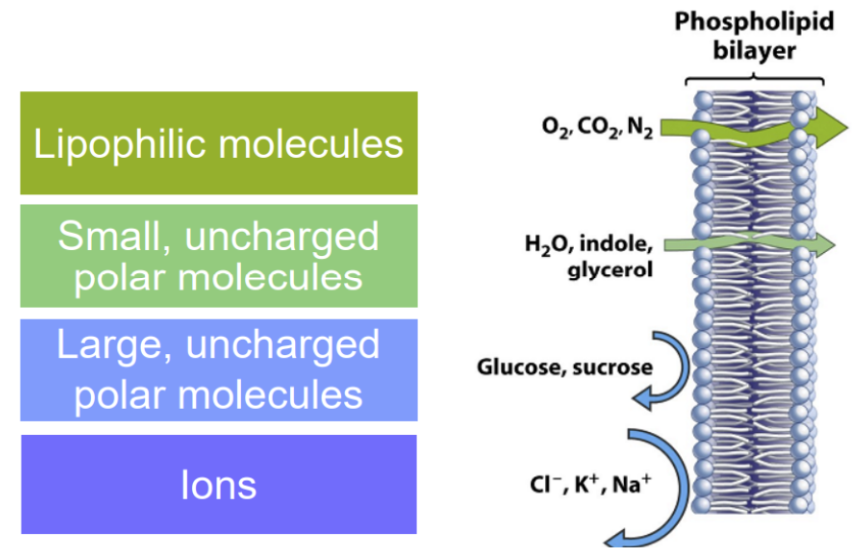
What is diffusion generally driven by?
a concentration gradient, going from high to low concentration (in order to spread out)
Fick’s diffusion equation
describes drug transport across a membrane → predicts drug absorption rate
Diffusion is faster when…
Diffusion coefficient (D) is larger (e.g., smaller MW drugs)
Surface area (A) is larger (e.g., intestine compared to skin)
(C1 - C2) is larger (e.g., higher concentration or dose)
Thickness of barrier (h) is smaller (e.g., intestine compared to skin)

What is Fick’s diffusion equation missing?
the cell membrane’s preference for lipophilic molecules
What is diffusion truly driven by?
membrane partitioning
Creates a true concentration gradient for diffusion
Partition coefficient (KD) consists of a lipophilic numerator and hydrophilic denominator
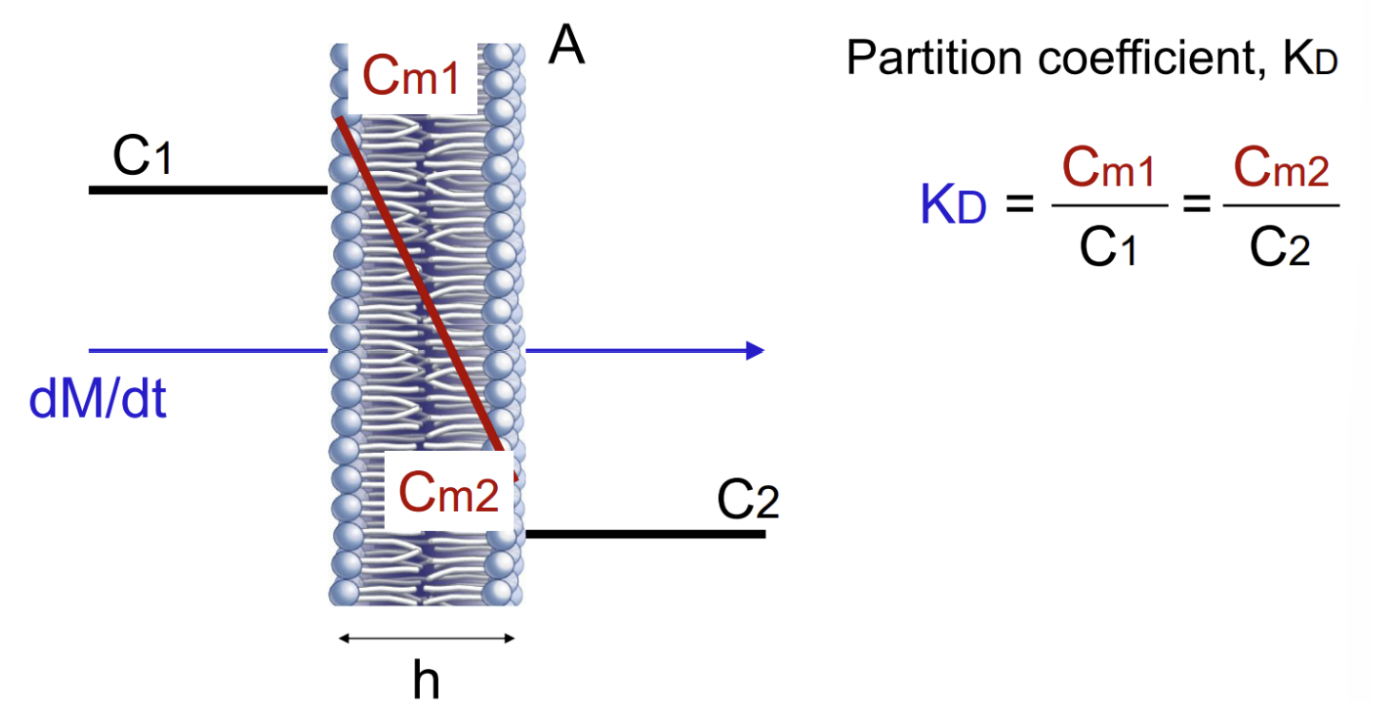
modified Fick’s equation to take into account membrane partitioning and lipophilicity
D is larger (e.g., small MW drugs > large MW drugs)
Kd is larger (e.g., lipophilic drugs > hydrophilic drugs)
A is larger
(C1 - C2) is larger
h is smaller
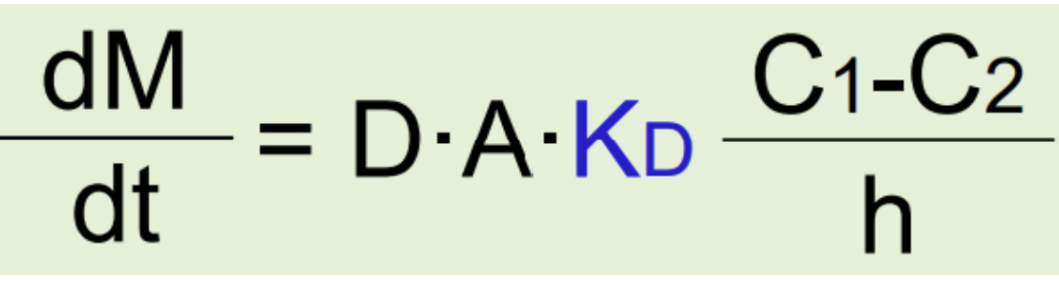
Ultimately, what sort of properties of a drug make it have the fastest diffuson and absorption?
Small MW
High lipophilicity (KD)
However, as lipophilicity (KD) increases, the rate of diffusion and absorption increases AS LONG AS…?
the drug is fully dissolved
If not, you can’t define a meaningful concentration (C) of the drug and for the gradient
When drugs are too lipophilic, they won’t get dissolved in water… so that is a big issue when designing for chemists is that you need enough lipophilicity to get past the cell membrane but not too much that it won’t get dissolved
diffusion
mass transfer of a dissolved substance from a region of high concentration to a region of low concentration in a single-phase system or across a permeable membrane
Stokes-Einstein equation
defines the diffusion coeffcient (D) in Fick’s diffusion equation and relates it to its attributes
D is defined by the solute size, the medium’s viscosity, and the medium’s temperature
Smaller size → Larger D → Greater diffusion and absorption
Lower viscosity → Larger D → Greater diffusion and absorption
Higher temperature → Larger D → Greater diffusion and absorption

pH-partition hypothesis
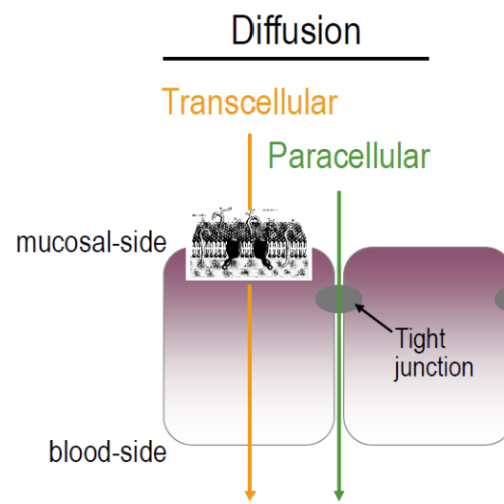
Only unionized, lipophilic (small MW) drugs can penetrate the cell membrane using trans(intra)-cellular diffusion, while ionized species are repelled
The fraction of unionized drugs at the absorption site determines their diffusive permeation across cell membranes
The Henderson-Hasselbalch’s principle allows estimation of a drug’s ionization/unionization state from its pKa
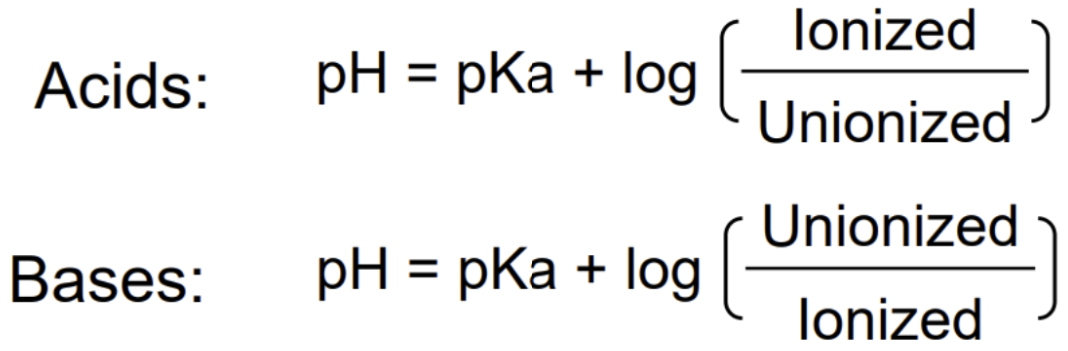
ionization effect on absorption
Bottom line: More % unionized = Higher diffusion rate and % absorption
What is a way hydrophilic, ionized (small MW) drugs can be absorbed then?
para(inter)-cellular diffusion (minor contribution)
Occurs mainly in the small intestine
Tight junctions filled with extracellular fluid within cellular membranes provide a diffusion pathway for hydrophilic and/or ionized drugs
However, the tight junction needs to be wide enough for large molecules. Otherwise, we have steric hindrance, resulting in either absorption getting hindered or not possible
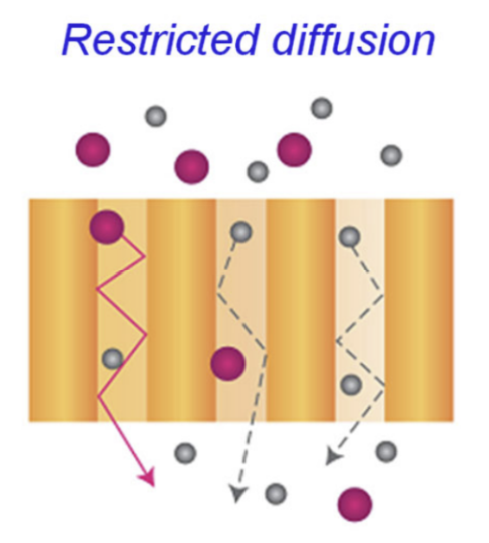
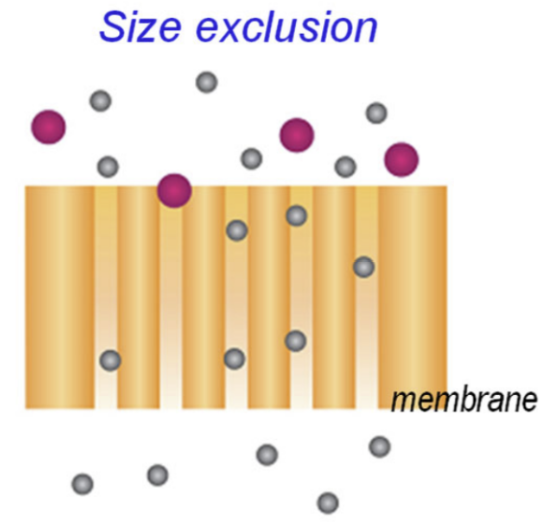
types of para(inter)-cellular diffusion
Restricted diffusion (problem with the pore)
Size exclusion (problem with the molecule size)
restricted diffusion
decrease in a particle's movement due to limited space or confined geometry, slowing down diffusion without changing the particle's size
size exclusion
What is the size cutoff?
separation principle where larger molecules are completely excluded from porous media (= ZERO absorption)
Size cutoff: 600 Da
rule of thumb for questions asking which type of diffusion (transcellular or paracellular) a drug will use
For acids:
If put into a more acidic environment → Unionized
If put into a more basic environment → Ionized
For bases:
If put in a more acidic environment → Ionized
If put in a more basic environment → Unionized
Cutoff for paracellular diffusion: 600 Da (i.e., poor/insignificant absorption)
With lipid membranes, cells are naturally capable of taking in lipophilic molecules and repelling hydrophilic molecules. However, what mechanism is available then if we need to take in essential hydrophilic molecules and ALSO remove lipophilic ones?
Transporters!
diffusion versus transporters
Most drug absorption occurs via diffusion
Transporter-mediated absorption, which is occurring in parallel with diffusion, is critical for certain orally administered drugs
specialized transport mechanisms
Uptake transporters
Efflux transporters
Cytosis
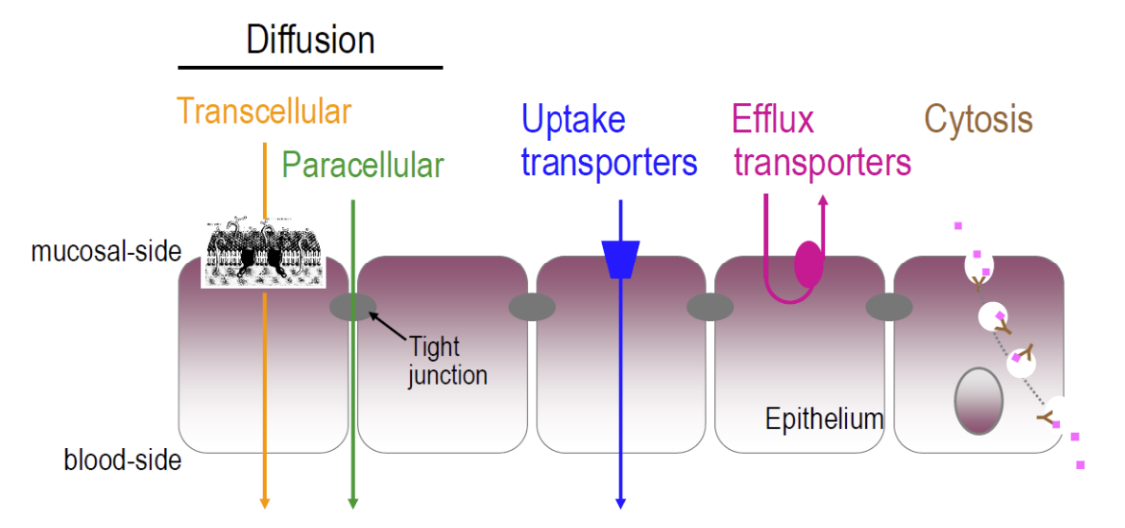
SLC uptake transporters
Types?
SoLute Carrier (SLC) protein-mediated uptake transport of substrate molecules upon binding, increasing absorption
Three different types:
Uniport: only one molecule needs to bind
Symport (cotransport system): two molecules need to bind on the same side and move in the same direction at the same time
Antiport: two molecules need to bind on opposite sides and move in different directions at the same time
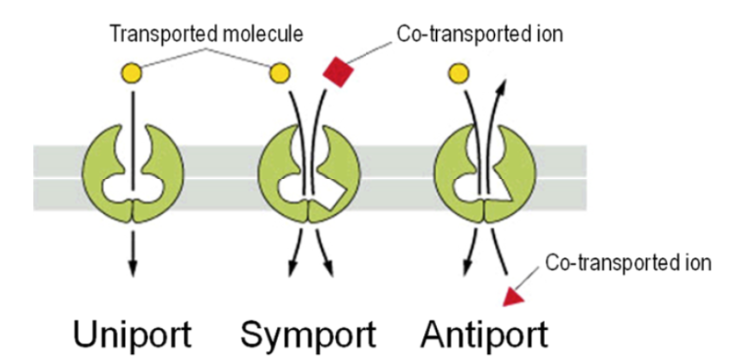
uniport SLC uptake transporter
Active or passive?
Is cell energy required?
Transport rate
Does structure matter?
Substrates (like vitamins and glucose) are passively transported into the cells via uniport proteins, following the concentration gradient
Cell energy is not required
The transport rate is…
Significantly greater than that predicted by diffusion (diffusion’s is arguably near 0 because it can’t take on hydrophilic molecules)
Nonlinear (unlike diffusion)
Not proportional to the concentration (dose) at all levels; it saturates at higher concentrations (doses)
Structure-selective and inhibited by chemically similar molecules
symport/antiport SLC uptake transporter
Active or passive?
Is cell energy required?
Transport rate
Does structure matter?
Substrates are actively transported into the cells via membrane proteins against a concentration gradient
Cellular ATP hydrolysis energy is used indirectly
The transport rate is nonlinear (unlike diffusion) and saturable
Transport rate is not proportional to the concentration (dose) at all levels
It saturates at higher concentrations (doses)
Structure-selective and inhibited by chemically similar molecules
examples of uptake transporters
PEPT1 (symporter that uses active transport)
OATP (uses active transport)
ABC efflux transporters
Active or passive?
Is cell energy required?
Transport rate
Does structure matter?
ATP-Binding Cassette (ABC) protein-mediated efflux transport of substrate molecules upon binding
Substrates are actively removed out of the cells against the concentration gradient, reducing intracellular concentration
Cellular ATP hydrolysis energy is used directly
The transport rate is nonlinear and saturable
Structure-selective and inhibited by chemically similar molecules
example of ABC efflux transporter
P-glycoprotein (PgP): Multi-Drug Resistance (MDR) efflux protein expressed in the intestinal epithelium, hepatocytes, renal proximal tubular cells, and brain capillary endothelium
Many tumor cells express this protein in order to protect themselves
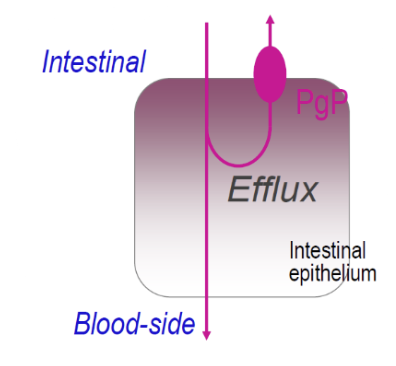
How does PgP affect gastrointestinal absorption?
Substrate drugs readily enter the cells due to their lipophilic nature
PgP pumps drug out of the cell back into the intestinal lumen
Net absorption is reduced, resulting in reduced blood levels
“selectivity” of PgP
PgP has many recognition sites, allowing for many molecules to be recognized. Chemically diverse drugs from different classes can act as substrates, including:
HIV protease inhibitors (e.g., rifampin)
Quinidine (antiarrhythmic)
Besides certain drugs—excipients, diets, and supplements can act as PgP inducers or inhibitors
Inducing PgP (e.g., rifampin) → Decreased levels of drug in blood
Inhibiting PgP (e.g., quinidine, grapefruit juice) → Increased levels of drug in blood
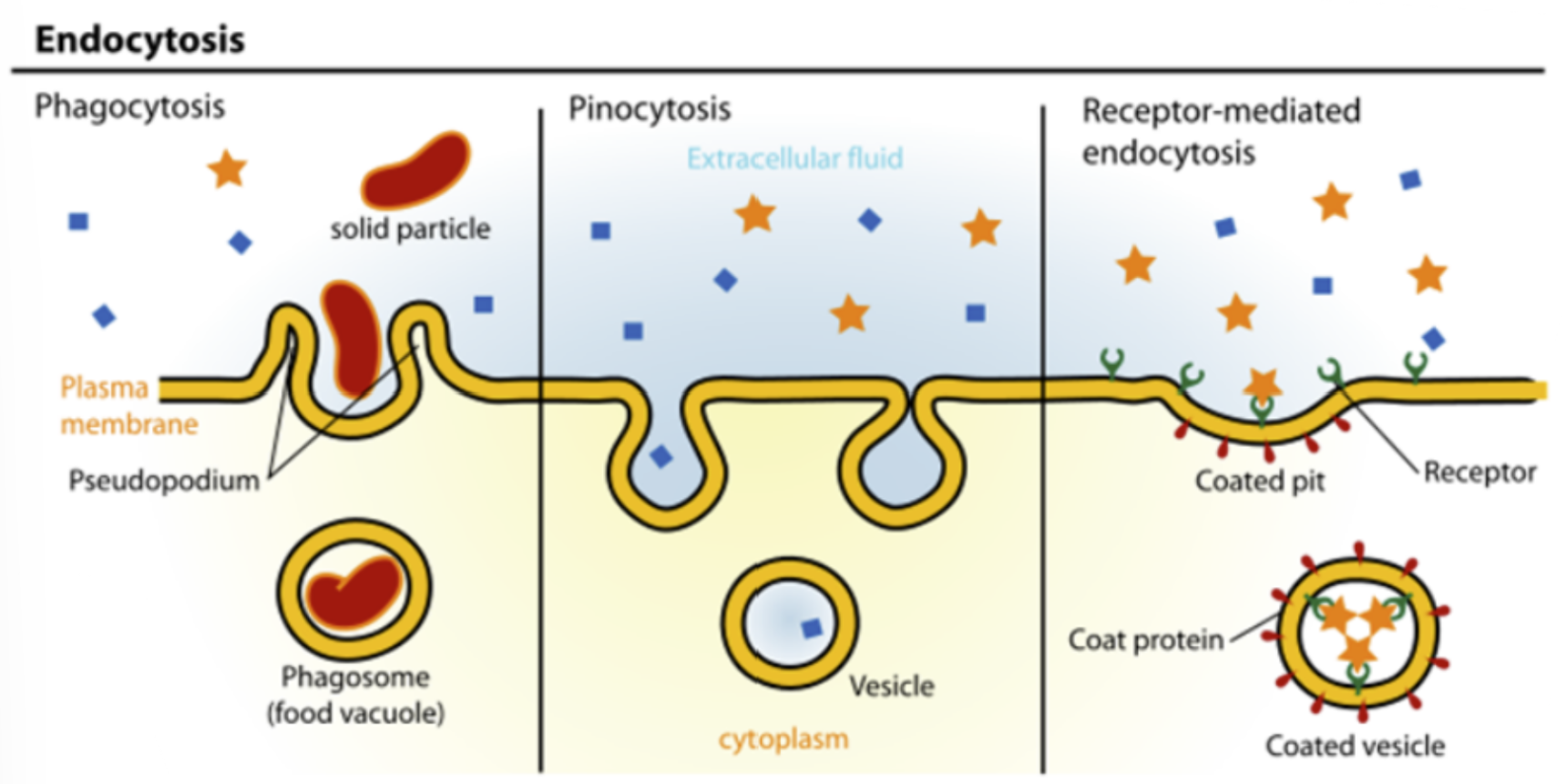
endocytosis/transcytosis
Cellular transport via phagocytosis and pinocytosis for certain macromolecules (e.g., insulin), fluids, and particulates
Mechanisms are similar to those for symport/antiport. However, membrane proteins are internalized
Example: Absorption of oral vaccine and transport of antibodies
Note: Small drugs are not absorbed by this mechanism
The fraction of drug that gets absorbed is described as…?
bioavailability
Dosage forms have been defined by what regulatory agency(ies)?
USP and FDA
Additives and excipients have been defined and approved by what regulatory agency(ies)?
FDA
What are the various oral dosage forms?
Liquid: Solution, syrup, elixir, tincture, suspension, and emulsion
The FDA discourages high-alcohol content in new, non-prescription oral products, which can affect the formulation of your traditional elixirs and tinctures
Solid: Powder, granule, capsule, tablet, effervescent tablet (EVT), orally-disintegrating tablet (ODT), enteric-coated (EC) and extended-release (ER) dosage form
Liquid in solid: Soft capsule, liquid gel
(Buccal/sublingual: Tablet, spray, lozenge, and gum)
mouth and esophagus
Type of epithelium?
Transit time? What does it depend on?
Luminal fluid pH?
Does drug dissolution and absorption occur?
Stratified squamous epithelium
Transit time: 10-14 seconds
Dosage form size- and shape-independent
Posture-dependent (slower in a supine position than in an upright position)
Luminal fluid has a pH of 5-7 (small buffer capacity)
Drug dissolution and absorption are insignificant because of…
The type of epithelium
The muscle they’re covered in
The quick transit time
What is a complication that can occur in the esophagus with drugs?
Can reach up to 20% when tablets and capsules are taken with little or no water
Higher in elderly populations
Adhesion and/or slower transit can cause:
Delayed drug appearance in the stomach and intestine, which can delay and/or reduce absorption (i.e., increased Tmax, decreased Cmax, and decreased AUC for acidic drugs)
Local esophageal damage (e.g., with NSAIDs)
The FDA recommends taking a full glass (> 250 mL) of water to not only help with dissolving of the drug, but also prevent adhesion of dosage forms
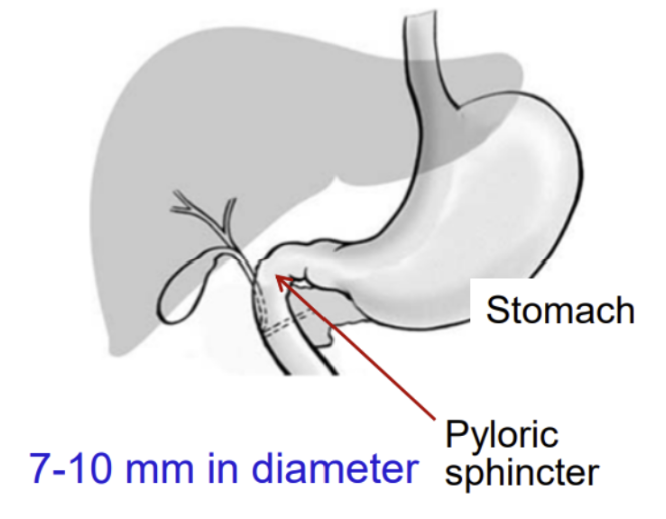
stomach
Type of epithelium?
Size of pyloric sphincter and what it entails?
Luminal fluid pH?
Does drug dissolution and absorption occur?
What does GI absorption depend on?
Simple columnar epithelium
Thinner (more permeable) than the membrane of the esophagus
Thicker (less permeable) than that of the small intestine
Pyloric sphincter is only 7-10 mm in diameter, so drugs need to be small enough or dissolved enough in order to pass through
1.0-1.5 L/day of acid secretion
Luminal fluid has a pH of 1.5-2.5 (almost no buffer capacity)
Food raises a pH to ~4.5, which can cause a change in the ionization state of the molecule and thus its absorption properties
Elderly populations often have an elevated pH
Here, unionized, small MW drugs are absorbed. Drugs that are not absorbed include:
Hydrophilic or ionized drugs
Large MW (> 600 Da) drugs
Peptide, protein, polysaccharide, and antibody drugs (must be administered via injection)
GI absorption depends on fat, calories, mass/volume, and temperature
How long does gastric emptying take? What does it depend on?
Gastric emptying is highly variable (1 to 7 hours), depending on dosage form, size, and food intake
Why do liquids leave the stomach faster than solids?
Liquids don’t require grinding/breakdown → faster gastric emptying → steeper disappearance slope and earlier peak concentration
How does dosage form affect time to peak concentration?
Liquids → faster peak (earlier Tmax)
Solids → slower peak (delayed Tmax)
How does fasting affect gastric transit time?
How do meals affect gastric transit time?
Fasted state → rapid transit (~1 hour) for both large and small pellets
Food increases transit time
Light meal → moderate delay
Heavy meal → significant delay
After meals, larger pellets empty slower than smaller pellets
What is the overall effect of food on drug absorption?
Delays gastric emptying, subsequently delaying transit to the small intestine, which delays Tmax
May or may not improve or have an impact on absorption (AUC) by allowing more time for drug breakdown/dissolution but also leaving the drug prone to secretions and gastric fluids that cause the concentration of the drug to decrease
The latter is especially a problem for acid-labile drugs
How does taking certain medications affect gastric emptying?
Some promote gastric emptying, which decreases Tmax
Others delay gastric emptying, which increases Tmax
summary on physiological gastric emptying
Highly variable (1 to 7 hours)
Liquid (fast) > Small solid > Large solid (slow)
Fasted (fast) > Fed (slow), which is fat- and mass-related
Other factors that promote gastric emptying: Anxiety, body position (lying on the right side), liquid intake, antiemetic drugs, and NaHCO3
Other factors that delay gastric emptying: Mental depression, body position (lying on the left side), ulcers, pyloric stenosis, and anticholinergics
gastric hydrolysis
How can we protect drugs from degradation?
Acid-labile drugs are quickly dissolved and degraded in the stomach
We can protect drugs using:
Enteric coating
Low-solubility salt (stearate)
Prodrug (ester)
Reducing particle size
small intestine
Type of epithelium?
What makes it unique?
Does drug dissolution and absorption occur here?
~6 m in length and arbitrarily divided into 3 segments (duodenum, jejunum, and ileum)
Simple columnar epithelium
A total surface area of 200 m2 due to epithelial folding, villi, and microvilli (so the surface area is not flat)
A majority of drugs are absorbed here via diffusion and transporters
How long is small intestine transit? What does it depend on?
Takes about 3 to 4 hours, irrespective of liquids or solids; large or small solids; or fed or fasted
A relatively short time window to complete absorption, but here is where most enzymatic degradation of drugs can occur (stomach only has pepsin)
Quite different from gastric emptying, which heavily depends on the presence of food
large intestine
Type of epithelium? Is it unique?
Luminal fluid pH?
Transit time? What does it depend on?
Does drug dissolution and absorption occur?
Simple columnar epithelium, but has no folds and villi, surface area is 3 m2
Luminal fluid pH = 5.5-7.8
Transit time: 15 hours (fasted) to 48 hours (fed)
i.e., depends on food intake
Drug absorption is limited in part by viscous and semisolid luminal contents; however, some drugs (e.g., theophylline and metoprolol) are well absorbed here in the large intestine. Furthermore, here lies the greatest water absorption and bacterial activity
What can a patient take to help improve dissolution and absorption of low-solubility or lipophilic drugs?
water
Enhances dissolution and absorption, but the extent is unpredictable
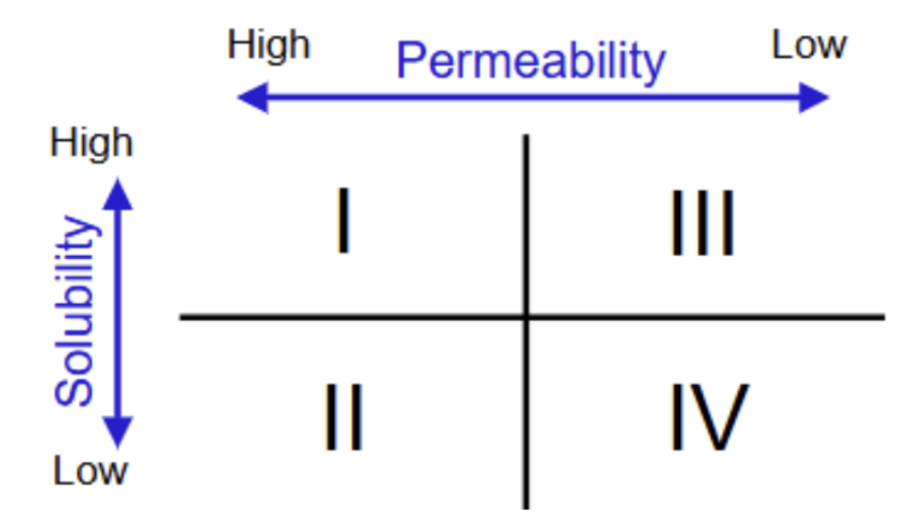
BCS guidance by the FDA
BCS = Biopharmaceutics Classification System
Framework for classifying drug substances based on aqueous solubility and intestinal permeability
process by which drugs undergo in the body, according to the BCS
When is a drug considered to have high solubility?
When is a drug considered to have high permeability?
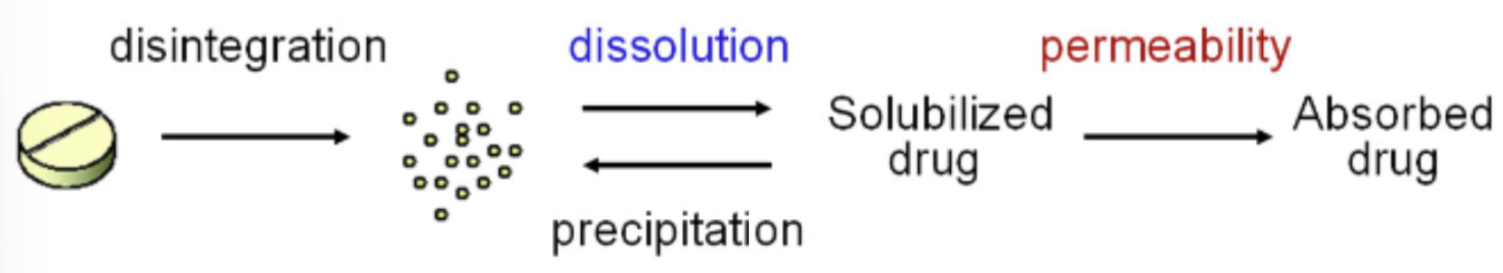
Disintegration (tablet breaking down into smaller particles in the stomach)
Dissolution (smaller particles are dissolved into solubilized drug)
High solubility: when the highest dose strength is soluble in 250 mL or less of aqueous media at pH = 1-7.5
Permeability (drug was able to be absorbed)
High permeability: when the extent of absorption in humans is determined to be 90% or more of the administered dose
Rank dosage forms from fastest to slowest complete dissolution and gastric emptying.
Solution, syrup, elixir
Powder, granule, EVT, ODT
Suspension, emulsion
Capsule
Tablet
Coated tablet
Enteric-coated (EC), extended-release (ER)
What can happen to a drug after GI absorption before reaching systemic circulation?
“competing losses” or “post-absorption events”
Drug can enters the portal vein → goes to liver → may be metabolized (loss) before reaching plasma
Metal complexation forms insoluble complexes that decrease absorption (e.g., antacids with Mg(OH)2 or Al(OH)3 decrease Cmax and AUC by ~90%)
What determines how much drug reaches systemic circulation?
Amount absorbed - amount metabolized (especially in liver)
First-pass metabolism is hepatic metabolism of a drug after GI absorption but before reaching systemic circulation. Thus, this decreases bioavailability; however, it does not affect Tmax
High bioavailability is determined by all three of these factors: low first-pass metabolism + high GI absorption + minimal post-absorption events
This means that high GI absorption does not necessarily equate to high bioavailability, because there are other factors at play
What causes double-peak plasma concentration profiles?
Food after dosing
Enterohepatic recycling
Drug is excreted in bile → reabsorbed in intestine by bacterial enzymes → causes secondary peak
summary on how food affects drug absorption
Delays gastric emptying
Alters GI pH
Changes solubility
Causes drug-food interactions
Affects transporters
↑ viscosity, bile flow, blood flow
The effects, if any, are greatest when drugs are taken shortly after food intake
summary on factors affecting GI absorption
Physiologic factors
Membrane physiology
GI tract physiology
Physicochemical factors
pKa
Lipophilicity
MW
Dissolution (formulation factor as well)
advantages of oral solution
Disintegration, deaggregation, and dissolution are not required for absorption
Gastric emptying is faster than for solid dosage forms, and food intake causes only a small delay
→ Fastest absorption among oral dosage forms
Homogeneity in solution allows drug doses to be measured and administered by volume
→ Dose strength is expressed as concentration
PK of an acidic drug (solution versus solid)
Solutions enable faster and greater/equal absorption than solids
PK of a basic drug (solution versus solid)
Absorption only begins when reaching the small intestine, not in the stomach
Solutions enable slightly faster and equivalent absorption relative to solids
disadvantages of oral solution
Maintaining chemical and physical stability is more challenging than for solids; the shelf-life is shorter
Drug solubility can be a challenge in formulations with doses of 5 mL (1 teaspoonful) to 15 mL (1 tablespoonful)
Dose measurements by patients and caregivers can be inaccurate and variable
Bulkier and less portable than solids
Drugs may precipitate in the GI tract as water intake is not instructed, which affects Cmax but does not affect Tmax
types of oral solution
Ready-to-use oral solution
Dry powder mixtures/granules for oral solution
Reconstitution by pharmacists (product shelf-life is shorter after reconstitution)
Less popular than suspension
Syrup, elixir, and tincture
ready-to-use oral solution
Stable in solution with R.T. storage
Preservatives and sweetening agents are added
Some sweetening agents, like glycerin, can also help improve solubility
syrup
Medicated?
Sucrose?
Ingredients?
A typical dose is how much?
concentrated aqueous solutions of sugar or sugar substitute with or without flavorant (usually viscous)
Medicated or non-medicated
Non-medicated syrups (e.g., Syrup NF: 85% sucrose in purified water) are used to prepare medicated syrups
Sucrose- or non-sucrose-based
60-80% sucrose for sweetness and viscosity
Artificial sugars (e.g., sorbitol, saccharin, aspartame, and maltitol) provide sweetness but lack viscosity. So, cellulose-based viscosity increasing agents (e.g., MC and HPMC) are also needed
Syrup ingredients:
API
Purified water
Stabilizer
Sucrose or artificial sugar + viscosity increasing agent
Preservative
Flavorant and colorant
Note: By definition (but not the case in reality), nonaqueous solvents (e.g., alcohol) are not used, and the formulation is viscous (i.e., 100% aqueous, viscous, and sweet)
A typical dose of volume is a tablespoonful
advantages of syrup dosage form
Use and acceptance by patients (especially pediatric and elderly), but not biopharmaceutics (When talking about PK, syrup had an earlier Tmax but a possibly smaller Cmax than tablet)
Does NOT need to be taken with water, as syrups are already aqueous, sweetened solutions meant to be taken as-is
elixir
Medicated?
Ingredients?
Comparison to syrups and suspensions?
clear, sweetened hydroalcoholic solutions, generally with flavorants
Medicated or non-medicated
Alcohol can be ethanol and/or sugar alcohol (e.g., glycerol, propylene glycol, and sorbitol)
Hydroalcohol dissolve both water- and alcohol-soluble ingredients (easier to prepare solutions than syrups)
Less sweet and less viscous than syrup
Less popular as suspensions gain popularity
non-medicated elixir
For extemporaneous filling of a prescription…
As a pleasant-tasting solvent for powder drugs
For dilution of medicated elixirs
Alcohol % can be low (8-10%), intermediate (25%), or high (75-78%)
Sweetening agents and colorants are added
Be aware of solubility and stability when mixing!
Alcohol concentration must be maintained
Chemical/physical compatibility must be ensured
Color and flavor must not have a conflict
medicated elixir
Commercial products contain alcohol (ethanol) by 5-20%
Those with ≥ 10% alcohol are self-preserving (i.e., no need to add preservatives)
A typical dose of volume is a “teaspoonful” or 5 mL
Generally not for children
tincture
alcoholic or hydroalcoholic extracts or solutions of substances (e.g., drugs)
Alcohol % can be 25-80%; many commercial products contain 45% or more
Tight closure to prevent evaporation loss
Mixing requires caution because of the higher concentration of alcohol
Storage should avoid heat/high temperatures and ignition
Very limited availability, but some USP products are popularly used (e.g., Paregoric USP, Opium USP)
suspension
Should be 100% Purified Water, USP that gets used to reconstitute and suspend the drug
Two types:
Ready-to-use oral suspension
Dry powder mixtures/granules for oral suspension
Reconstitution by pharmacists (product shelf-life is shorter after reconstitution)
Used to be a compromise of solutions, but now has become a preferable option, even over solids
Popular APIs: Antibiotics, antifungals, and antacids
pros of oral suspensions
Dissolution can be faster than for solids
Gastric emptying is faster than for solid dosage forms, and food intake causes only a small delay → Absorption and onset is faster or equivalent compared to solids
Drugs can be chemically stable, with a longer shelf-life compared to solutions
cons of oral suspensions
Formulation is challenging for water-soluble drugs (they often end up with a solution instead)
Dose measurement and physical size present practical disadvantages, like solutions
If the liquid is relatively heavy and bulky, that creates higher transportation costs
When comparing the PK of different dosage forms, what should you take note of?
Tmax is directly comparable
Cmax requires the dose to be equivalent before comparisons can be made
emulsion
oil-in-water (o/w) disperse systems with emulsifying agents
As of today, we do not have any emulsion dosage forms that contain the APIs you typically think of. Pharmacological agents are not formulated; the oil itself is the active ingredient!
Currently used only for local GI effects, not for systemic absorption or action
advantages of oral solid dosage forms
Natural and pain-free
Simple, small, and convenient
Accurate dose (i.e., essentially already measured for the patient when it’s a tablet)
Exception: Some laxative products requires the patient to scoop out the current amount of powder
Increased stability and longer shelf-life
Relatively inexpensive
disadvantages of oral solid dosage forms (compared to liquid)
Absorption and onset of action are slower because it takes a while to disintegrate and dissolve
Absorption is more influenced by food intake
types of oral solid dosage forms
What do all solid dosage forms require?
Powders and granules
Capsules
Hard and soft
Tablets
Uncoated and coated
Orally-disintegrating tablets (ODTs)
Effervescent tablets (EVTs) (not directly placed into the mouth; must be placed in a glass of water or tea and drunk all together, which makes the body process this in a similar way to a suspension)
Enteric-coated (EC) tablets
Extended-release (ER) tablets
(All solid dosage forms require nice powder formulations and to also be taken with water except for some ODTs!)
diluent
an essential excipient to increase bulk, compressibility, and homogeneity so that patients have something already accurately measured and can be physically handled
Enables accurate handling and administration of drugs in a small quantity
Other names: Filler, bulking agent
powders
finely divided drug particles
Fast dissolution → Fast absorption
Flexible dosing (can adjust amount easily)
Maximum tablet mass: ~2 g (otherwise, the patient can’t swallow it)
Drug stability in solution does not need to be ensured (since the patient is reconstituting it)
However, there are often issues with flow, making it hard to handle/manufacture
Content uniformity must be ensured (i.e., no matter where the powder is scooped, you will have the same amount of drug in it proportionally), but powders often segregate due to differences in particle size, shape, and density. What is a possible solution?
turning the drug particles into granules
granules
2-5 mm irregular agglomerates of small particles, prepared by wet or dry methods
Unlike/relative to powders:
Each agglomerate contains all the ingredients
Better content uniformity
Larger in size, enabling better flow properties in manufacturing, but requires an extra processing step
Higher in density, which allows smaller dosage forms and reduced storage space
Slightly slower dissolution
types of powders and granules
Bulk powders
Single dose-unit powders
How should oral powders and granules be administered?
must be added to water first and then drunk, not just taking the powder directly into the mouth
Behaves like a suspension in our system
Don’t need to worry about ensuring the stability because the patient “reconstituted” it themselves and took it immediately after; however, there is a concern of inaccurate measuring of doses
advantages of powders and granules
Easier in swallowing with water intake
High drug doses can be formulated (e.g., > 2 g)
It is really difficult to fill 2 g of a drug into a capsule or compress into a tablet because it’s just too large
Quick dispersion and dissolution → Fast absorption
Faster gastric emptying (less influenced by food intake) than capsules/tablets
disadvantages of powders and granules
Bulk powders are less convenient for carrying/storage
Acid-labile or hygroscopic drugs are unsuitable for this dosage form
Acid-labile drugs dissolve and degrade quickly at the stomach’s pH, which can reduce their effectiveness significantly
Hygroscopic drugs absorb water from their environment, leading to poor flowability, caking, instability, and degradation. They become moist clumps that makes them sticky, causing processing issues and inconsistent, inaccurate dosing
two types of capsules
Powders/granules in a hard shell
Liquid in a soft gelatin shell (→ has a layer of protection, but possible barrier for dissolution)
gelatin capsule ingredients
Gelatin: main ingredient; animal-derived; soluble at 37 °C in 10-60 min (i.e., not instantaneous, delaying onset of action)
Water: to provide flexibility and maintain 13-16% moisture
Preservative (e.g., parabens)
Plasticizer: used only in soft capsules for sealing (e.g., glycerin, sorbitol)
preservative
prevent microbial growth that could result from using water as an ingredient for either the powder or the coating (if applicable)
innovation in capsules
Hydroxylpropyl methylcellulose (HPMC) capsule
Pullulan capsule
hydroxylpropyl methylcellulose (HPMC) capsule
Made up of?
Compared to gelatin capsules…
Tidbit
Plant-derived, suitable for vegetarians/vegans
Compared to gelatin capsules…
More stable, and less sensitive to temperature or humidity
Lower in water content (4-6%), often allowing no use of preservatives
More resistant to moisture sorption
Slower in disintegration
More expensive
Tidbit: Used in approved oral products (e.g., Talzenna)