Quantitative analysis
1/19
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
20 Terms
What information can be obtained about receptors?
The affinity of drug-receptor interactions
The number of binding sites —> need to be able to spot additional binding sites to interpret data correctly
Pharmacological properties
Structure function relationships derived from pharmacological profiles
However, things like radioligand studies cannot give us information about receptor efficacy
How?
Incubate the tissues, cells etc with radiolabelled ligand
Separate the free drug from the bound drug by centrifuge or filtration (most of the time) because there will be no difference in the signal of a radioligand whether bound or free. With fluorescence we may be able to just look at the signal from the bound ligand without separating the free ligand
Estimate the amount bound at different concentrations of ligand
Keep the amount of receptor constant but vary the amount of ligand to try and vary this profile
Use other drugs to displace the ligand to characterise the pharmacology
Introduce mutations into receptor protein to investigate structure function
Interpreting results
Total ligand binding curve will not saturate because this contains specific and non-specific ligand binding (ligand sticking to plastic ware or embedded in memb)
Specific binding will saturate and will be a rectangular hyperbole shape which will also give us info about the affinity (Kd) and receptor density (Bmax)
Lowest Kd will be the tightest binding
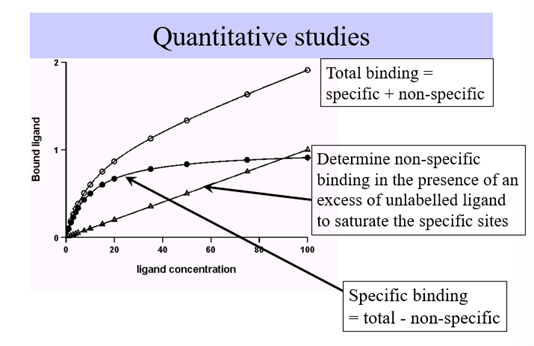
Why bother with quantitative analysis
Pharmacological profiling (can separate ligand binding for receptor subtypes)
Identification and isolation of receptors
Quantifying receptor number
Analysis of binding data
Assume:
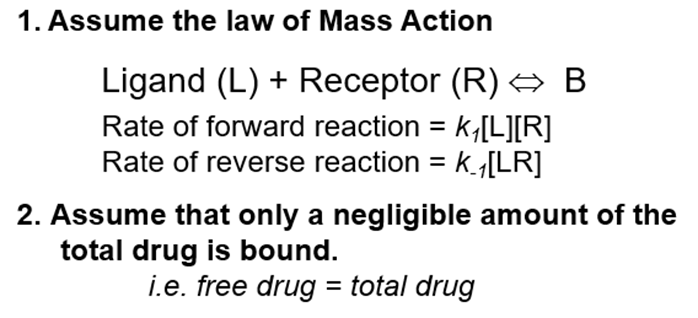
Once we’ve reached equilibrium, the rate of the forward reaction is equal to the rate of the reverse reaction k1[L][R] = k-1[B]
Because we use such an excess of ligand on a very small concentration of receptors we are able to make the second assumption
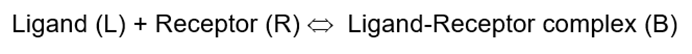
Equilibrium constants
We use disassociation constant rather than association constant (Ka)
Equilibrium association rate = k1/k-1 = Ka, dissociation rate constant = k-1/k1
The reason that we chose kD because kA has units of litres per mole while kd has units of mol/L (molar) which is easier to understand and relate bac to our ligand binding experiments

At equilibrium
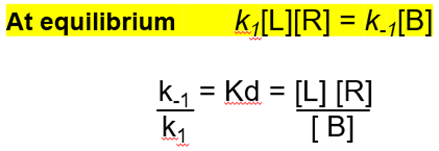
Equilibrium constants to measure affinity
On the right is is we think of Kd at equilibrium whereas on the right is in terms of rate constant
The issue is that we don’t often know the concentration of receptor to calculate the kD
So in order to take out the term [R] we can substitute this for Bmax – [B]
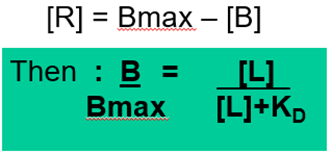
This equation applies for simple bimolecular interactions between a ligand and its receptor
Plotting ligand concentrations against bound ligand
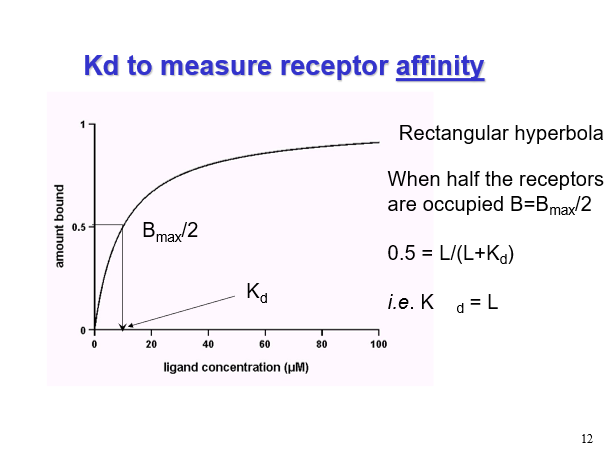
Bmax
Gives us the total number of receptors that you’ve bound in that experiment (total receptors)
Typically 10^-12 to 10^-15 moles/mg of protein which gives you a window to check for in your calculations
Specific activity
Is the amount of radioactivity of a particular radionucleide per mole of radioligand
So if we know the specific activity of a radioligand we can use this to sovert back to a concentration of ligand that must be bound
Example of data
Example is of H3 spiroperidol binding to pig striatal membranes
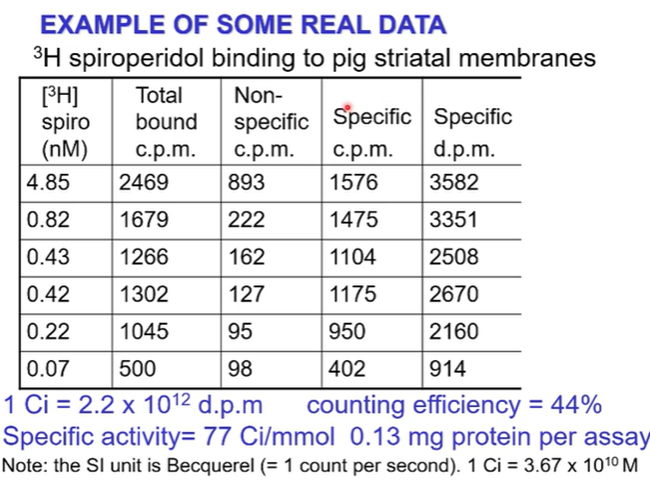
Column on the left is the radioligand in nM across a range of concentrations as you go down. You then separate out the bound from the free and measure the bound with a scintillation counter, recorded in the second column (counts per minute)
The experiment will then be repeated, flushing with non specific cold ligand to ascertain how much non-specific binding there is (recorded in the third column)
Now subtract the non specific from the total bound to have specific bound (fourth column) —> this is the data that we’re interested in but its still in cpm
The machine that we count radiation is not 100% efficient at recording all the radiation. We need to take the counting efficiency into account —> in this case is 44% so if there were 100 radioactive particles would only count 44 of them
To convert between dpm (disintegrations per minute) to cpm we multiply by the counting efficiency
Dpm is the actual amount of radiation being emitted per minute
With every ligand that is bound we get information about the specific activity in curies per mmol but our data is in dpm (disintegrations per minute)
To covert between these 1 Ci (curies) = 2.2 x10^12 dpm
How to find Kd and Bmax
Will always be told the conversion between curies and dpm, the specific activity in ci/mmol and the amount of protein er assay.

Summary of working out ligand binding
Add varying concentrations of radioligand
Record total bound in scintillation counter which gives counts in cpm
Then flood with cold ligand to assess non-specific binding and subtract that from the total to give you specific binding
Then account for the counting efficiency of the machine (cpm→dpm)
E.g: if this is 44% then divide by 44 and x100 to go from cpm to dpm
Convert our dpm value to curies (1Ci Is 2.2x12^12 dpm)
We are given the specific activity of the ligand in curies per mole (Ci/mmol) so will have to work out how much protein is bound based off the number of curies emitted by the radioligand
Specific activity is in Curies/mmol so convert to to curies/mol by x1000
Dpm/ (2.2x10^12 x specific activity in mol) = moles of ligand bound
Moles of ligand bound/milligrams of protein to give us femtomoles per milligram of protein
Plotting data
Direct plot
Bound ligand in femtomoles per milligram on the y axis, concentration of radioligand on the x axis 9 (nM)
We observe a rectangular hyperbole → this is known as a direct plot
Some of the benefits of the direct plot is that it doesn’t require any modelling, data transformation and there’s no distortion of data points
But one of the limitations is that to accurately calculate Bmax you need the receptor to be fully saturated which requires very high ligand concentrations (around 100x the Kd)
Scatchard plot
Easily linearise the data using the scatchard equation
Another method is the Lineweaver Burke plot but this is less favoured compared to the Scatchard plot
A plot of bound/free against bound should give us a straight line which has a slope of -1/kd and the x intercept should give us Bmax
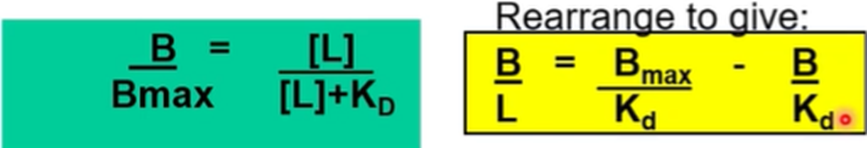
Will usually give us a line of best fit, not always perfect data
Can extrapolate this slope to reach the x axis
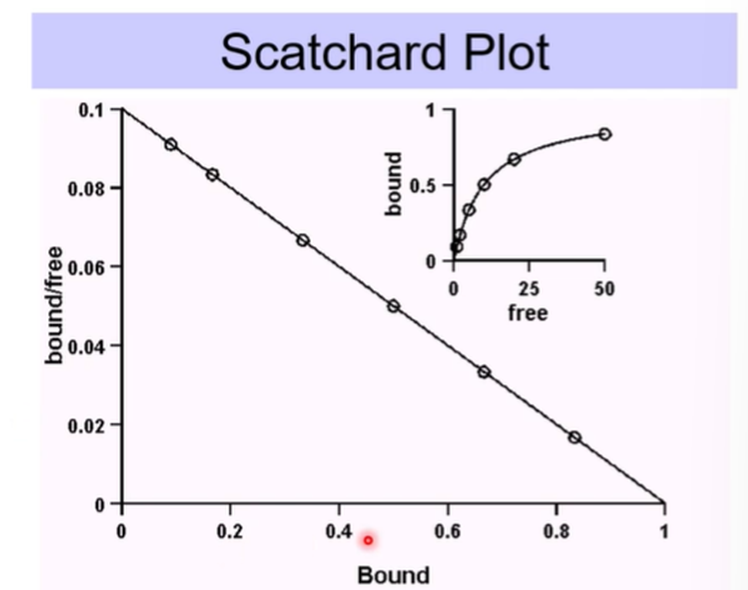
What if we don’t get a straight line?
If our scatchard plot is slightly curved might indicate
more than one binding site (site heterogeneity)
Negatve cooperativity
The curve should have two distinct phases:
At low concentration a slightly linear portion
At high concentration of free ligand a second linear portion with a different gradient
At low occupancy, the ligand will bind to the low-affinity site but at higher concentration there’s a second site with a weaker affinity which is occupied
We would not be able to interpret this from a direct plot
To calculate Bmax we draw a tangent and extract a Bmax for both portions and then add them together
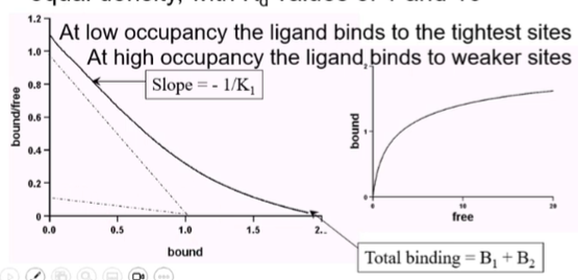
How deformed does it have to be?
Depends on the difference in affinities of the two binding sites → binding sites with small differences in affinity will be less deformed
Negative cooperativity
Can also be indicated from a bent looking Scatchard plot because binding of the ligand induces a conformational change which alters the affinity
Typically an agonist
Rarely see positive cooperativity in receptors
Hill plot
If we suspect that there may be site heterogeneity or cooperativity then we can do the Hill analysis
Can tell us about the number of binding sites and whether they display cooperativity
To plot this then we would plot log[B/Bmax-B] against logL which will give us a straight line with the slope of n which is the Hill coefficient
If we have a single site with a kd of 1 then we will have a hill slope of 1
If we have a hill slope less than 1 it is indicative of site heterogeneity
The smaller the hill slope is the greater the difference is between the Kd of the binding sites

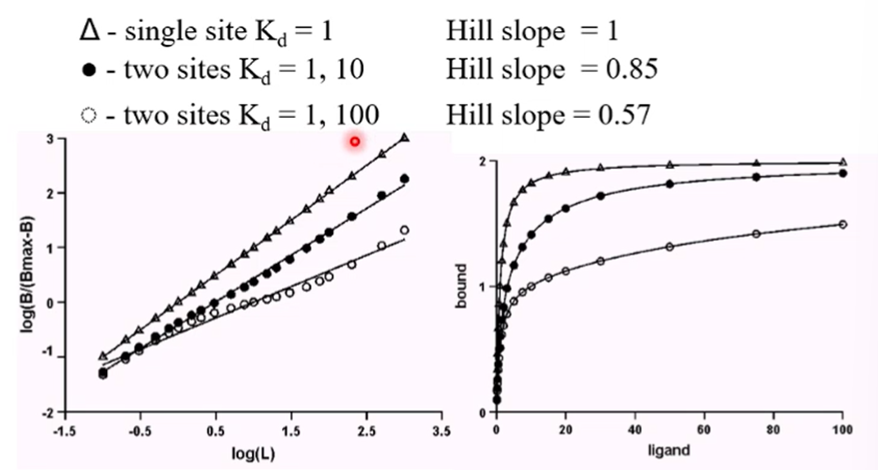
Displacement curves
Can use displacement curves to charcaterise a panel of other ligands
Have a fixed concentration of radioligands in the assay and can displace this with different amounts of an unlabelled ligand
Plot log of unlabelled ligand on the x and amount of ligand bound on the y
May not always reach zero
Want to know the conc of ligand that displaced half of the bound ligand which gives us an Ic50 of the second unknown ligand
Useful if we are unable to radiolabel the radioligand under investigation
IC50 is dependent on the concentration fo the labelled ligand and its Kd
Can then use the Cheung prusoff equation which corrects for this and calculates the equilibrium dissociation constant for the inhibitor Ki
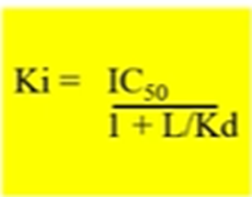
Bottom half of the equation relates to the radioligand concentration and the radioligand Kd
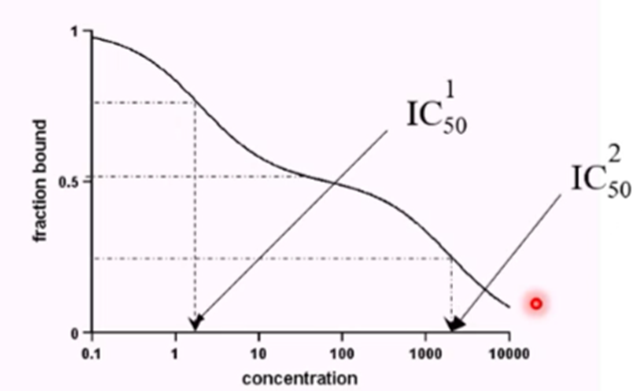
Displacement curves can also show the presence of multiple binding sites
E.g if there are 2 receptor sites to which the radiolabelled ligand binds with equal affinity, but which have different dissociation constants then they will be displaced at different conecntrations
Can observe two humps in our displacement curve and we have to raed off 2 IC50 values (sometimes may not be well resolved if the Kd is similar)
If we are still unsure of whether the displacement curve shows site heterogeneity then we can always do a hill plot analysis to determine this
Cloned receptors
If we clone receptors and express them in a given cell model, sometimes there will be G proteins that these cloned receptors couple to that they wouldn’t normally do
If this is the case, sometimes if we trigger a conformational change then we trigger other downstream effects → this can make our displacement curve shallower
Biological context of the experiment is important
Example: looking at beta receptors and looking for agonists often measured by the displacement of 125I cyanopondolol → Displacement curves showing single binding sites in cells lacking the G protein but show complex binding curves in cells which express the G protein