3: metode za določanje 3D strukture
1/32
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
33 Terms
x-žarkovna kristalografija
1. metoda za določanje strukture (mioglobin 1958)
rabimo kristal molekule (proteina) → obsevamo ga z x-žarki (val dol 1-2A) → ko žarki interagirajo z elektroni v kristalu, pride do sipanja in interference - elektroni absorbirajo fotone iz žarkov in oddajo fotone z enako energijo → pod določenimi koti pride do interference med oddanimi (sipanimi) žarki → lahko izmerimo intenziteto in izračunamo elektronsko gostoto → vanjo zgradimo model strukture
zakaj x-žarki in kristali
žarki: najmanjša stvar, ki jo lahko razločimo je velikosti λ/2, zato če hočemo na nivoju A gledat rabimo čim manjšo λ žarkov (x-žarki so to)
kristali: nemoremo dat proteina pod mikroskop ker za x-žarke nimamo leč + ena molekula sipa žarke zelo šibko, zato jih damo več skupi (v kristal)
vir x-žarkov
katodna cev: katoda oddaja elektrone, v el polju se pospešijo → pičijo proti anodi, kjer je tarča - segrevanje → 1% energije se izseva kot x-žarki
sinhrotron: produciramo elektrone, ki jih nato pospešimo (skoraj do svetlobne hitrosti), šibajo po obroču - pot jim ukrivimo z magneti → pri zaviranju izsevajo fotone (v različnih žarkovnih linijah)
sinhrotron>katodna cev, ker imamo veliko več fotonov, večjo briljantnost, lahko izberemo val. dol.
kristalizacija proteinov
kristal more bit >99% čist in zelo homogen (ista konformacija, PTM)
da pride do kristalizacije moramo zmanjšati topnost proteina - dodamo obarjalna sredstva (lahko pride do denaturacije - nočemo, ali agregacije/oborine - also nočemo → odvisno od lastnosti proteina in raztopine + okolja)
proteinu dodamo shit, ki mu zmanjša topnost: npr izsoljevanje - pri malo soli je alright topen, z več soli je ful topen (ker se ioni soli vežejo na prot), pri ful soli skoraj ni topen (ker vso vodo vežejo ioni soli)
kristalizacija metode v praksi
difuzija vodne pare: kapljica z vzorcem + precipitantom je v zaprti posodi (viseča ali sedeča), zraven v posodi je raztopina precipitanta → iz kapljice hlapi voda v raztopino precipi, kjer je je manjša konc. (difuzija vodne pare) → vedno manj v kapljici → prot kristalizira
difuzija “preko meje”: v kapilari na eni strani vzorec prot, na drugi precipi → difuzija - precipi gre na stran prot ker ga je manj tam → manj vode v razt. proteina → kristali
“šaržne” metode: razt proteina zaprta s polprepustno membrano → precipi difundira iz rezervoarja v vzorec → kristal
kateri proteini bojo lažje kristalizirali
majhni
brez daljših fleksibilnih regij
nenabiti
lastnosti makromolekulskih kristalov
notr je tudi topilo (50-60%)
struktura proteinov se ne spremeni, encimi ostanejo katalitsko aktivni
zelo visoka koncentracija proteina - več kot 3x več kot v celici (700 mg/ml cca)
priprava kristalov na snemanje difrakcijskih podatkov
kapilara (pri sobni temp.) - da se ne izsuši, bolj verjetno poškodbe od sevanja + slabša ločljivost
zanka - če zamrznemo vzorec v lqN2 (100K, zamrznemo zelo na hitro da ni kristalov ledu) - boljša ločljivost
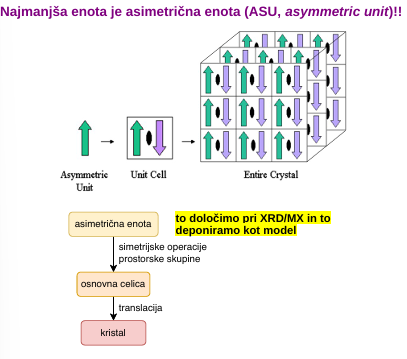
kristali in simetrija
v kristalu imajo molekule ponavljajoč se motiv = osnovna celica - najmanjšo možno os. cel. zberemo (je pa os cel zgolj konstrukt, ni nujno da je v eni celici ena cela molekula proteina
v kristalih lahko izvajamo simetrijske operacije (32 kombinacij) - npr rotacija 360/n, vijačna os (rotacija + translacija); in prostorske skupine
najmanjša enota je ASU - asimetrična enota → z simetrijskimi operacijami jih damo v osnovno celico → translacija → kristal
difrakcija
sipani žarki od vzorca grejo na vse strani → pod določenimi koti konstruktivna interferenca - dobimo difrakcijske točke (basically ojačitev signala je)
difrakcijski vzorec je signal, ki ga detektiramo → merimo intenziteto
na podlagi difrakcijskega vzorca se lahko izračuna elektronska gostota (= fourierjeva sinteza strukturnih faktorjev F)
faziranje
iz difrakcijskega vzorca izračunamo lahko strukturne faktorje, faz pa ne moremo določiti → lahko molekulska zamenjava - uporabimo že znano strukturo proteina, ki je podoben vzorcu in njegove faze damo v karto el gostote
gradnja strukture v elektronsko gostoto
poznamo ak zaporedje in imamo karto el gostote → lahko vanjo zgradimo model strukture (večja kot je ločljivost, lažje gradimo - več detajlov lahko prepoznamo)
pri gradnji modela upoštevamo, kar smo izmerili (el gostota), ampak more štimat tudi stereokemijskim parametrom - dolžina vezi, koti, polarnost, kontakti…
kako pilimo model
ciklično
imamo protein A, ki smo mu z XRD dobili strukturne faktorje → za faziranje uporabimo strukturo proteina B (znana struktura, podobno ak kot A) → zgradimo karto el gostote od A, notr je struktura od B → strukturo od B prilagodimo zaporedju in gostoti od A → dobimo novo strukturo → jo uporabimo za izračun nove boljše karte el gostote → notr je modified B struktura od prej → jo zopet prilagodimo in to ponavljamo, dokler nimamo modela strukture proteina A
validacija modela strukture
ko pilimo model nam faktor R pove koliko se model strukture ujema z eksperimentalnimi podatki (nižji kot je, bolj se ujema)
v PDB najdemo podatke o R faktorju za različna ujemanja
kaj je NCS
ne-kristalografska simetrija
v 1 asimetrični enoti je lahko več molekul proteina, ki pa se konformacijsko razlikujejo → ni kristalne simetrije
voda in vodik v kristalnih strukturah
Hja ne vidimo ker slabo sipa X-žarke (samo en elektron ima)
v kristalu je voda večinoma v kanalih med makromolekulami - naključno orientirana → ne vidimo
vidimo pa tisto vodo, ki je tik ob proteinu - tvori H-vezi s proteinom =kristalna voda
kaj pove zasedenost, kaj je B faktor
zasedenost: kolikšen delež ASU ima atom na tem mestu (če so različne konformacije prot, če ni liganda pri nekaterih potem zasedenost =/= 1)
B faktor: termična vibracija atoma - višji pri atomih v bolj fleksibilnih regijah (zanke)
Serijska femtosekundna kristalografija (SFX)
pri XRD težko dobimo velike kristale molekule, ne moremo opazovati dinamike → če uporabimo majhne kristale, slabo sipajo in se hitro uničijo
pri SFX pa lahko majhne kristale - naredimo 1 sliko na 1 kristal (rabimo jih dosti) → obsijemo z zelo intenzivnim pulzom žarkov - z laserjem (posnamemo difr. vzorec preden se kristal uniči)
lahko posnamemo reakcije - damo substrat v nanokristale ali pa sprožimo reakcijo z laserjem (zamik med laserjem in xžarki)
NMR spektroskopija
spektroskopska tehnika - gledamo absorpcijo in emisijo energije
potrebujemo vzorec z NMR aktivnimi jedri !
vzorec izpostavimo močnemu mag. polju → NMR jedra zavzamejo 1 od 2 stanj → gledamo prehode med stanji - odvisni so od kemijske okolice jedra - lahko jim pripišemo identiteto
priprava vzorcev za NMR
NMR aktivna jedra imajo spinsko kvantno število =/= 0: 1H, 13C, 15N, 2H, 14N)
pri majhnih molekulah dovolj 1H, pri večjih več različnih
15N: gojimo bakterije v gojišču z virom dušika 15NH4Cl
13C: edini vir ogljika 13C-glukoza
2H: gojenje v D2O
vzorec mora biti v raztopini
kako deluje ta shit
jedra imajo spin - vrtijo se okrog neke osi, spini so v vzorcu naključno orientirani → damo v magnetno polje → spini se orientirajo v 2 možni stanji - več v stanje z nižjo energ (razllika energ med stanjema sorazmerna z jakostjo mag polja) → ko v vzorec dovedemo energijo - radiofrekvenčni pulz, se pri določeni frekvenci energ absorbira (frekvenca ustreza razliki med stanjema) - prehod med stanjema → ko RF pulz izklopimo, se energija emitira → zaznavamo signal in dobimo NMR spekter
NMR spektri
kemijski premik - kako se resonančna frekvenca (RF) razlikuje od standardnega jedra zaradi drugačnega kem okolja
senčenje: elektronsko bogate regije senčijo jedra → nižja RF, elektronsko revne regije manj senčijo → višja RF
sklopitev med spini: bližnja neekvivalentna jedra vplivajo na energ nivoje drug drugega → multiplet v spektru
spektri pri strukturi proteinov
v proteinih je zelo zelo veliko atomov → se zelo malo razlikujejo nekateri → prekrivanje signalov (zato tudi nemoremo prevelikih proteinov)
zato so pri prot večdimenzionalni spektri (1H, 15N) in različne RF sekvence - da dobimo tudi druge sklopitve:
COSY - sklopitve med jedri atomov, ki so kovalentno vezani na druge atome → ugotovimo kateri ak ostanek je
NOE - sklopitve med jedri ki so prostorsko blizu → ugotovimo sekvenco
izračun strukture iz spektra
iz spektra lahko razberemo bližino NMR aktivnih jeder med sabo → prostorske omejitve + upoštevamo stereokemijske parametre
izvedemo energijsko minimizacijo
naredimo več modelov, ki se ujemajo s parametri in imajo čim nižjo energijo = ansambel modelov → vsi so deponirani v PDB
H atome vidimo v modelih
lahko proučujemo kar v celici, tudi dinamične dtrukture lahko
elektronska mikroskopija
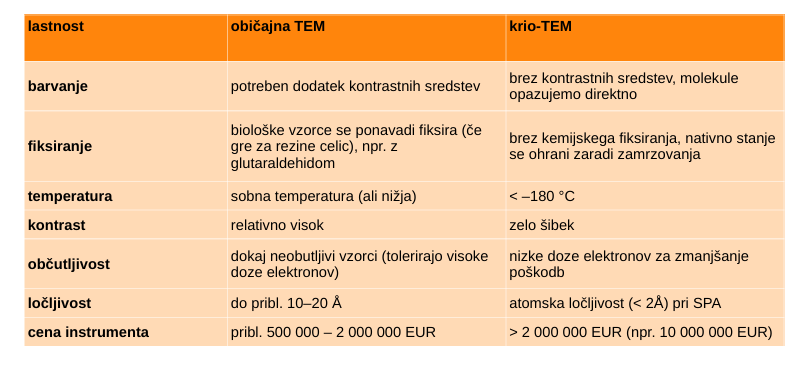
TEM verzija - presevna/transmisijska
vir elektronov → elektrone se pospeši, elektromagneti jih oblikujejo v curek → zadanejo vzorec → grejo skozi in se odklonijo glede na debelino in obliko vzorca → detekcija elektronov → slika
krio-EM
vzorec je zamrznjen na kovinski mrežici, prekriti z grafitom - ne rabimo fiksirat
krio EM vs EM (glej sliko)
negativno barvanje
molekule same imajo šibek kontrast → z depozicijo kovinskih atomov ga povečamo
vzorcu dodamo kovinske spojine (OsO4, U-spojine, amonijev molibdat…) → kjer je manj kovine - direkt na površini vzorca, elektroni ševedno lahko prehajajo → svetlejše; kjer je kovine več - ob vzorcu, težje prehajajo → temnejše
+ višji kontrast, easy priprava
- poškodba proteina, nižja ločljivost (10-20A), lahko nastanejo artefakti
krio-EM SPA
single particle analysis
vzorec na mrežico, se zelo na hitro zamrzne → zajem mikrografij na krio el mikroskopu → na mikrografijah detekcija posameznih delcev → klasifikacija njih v razrede → karta gostote glede na Coulombov potencial (elektrostatski potencial zaradi e-) → gradnja modela v karto gostote
vzorec mora biti zelo čist, homogen konformacijsko
rabimo ful dosti delcev zbrat skupi da dobimo strukturo
gradnja modela krioEM SPA
v obliko gostote prilegamo enote (posamezne ostanke, elemente sek strukt, domene, celotne proteine) → manjše delce kot prilegamo, boljša bo ločljivost
Q vrednost za podobnost vrednosti gostote karte okoli atoma vs teoretične gostote (1 je max)
krio-ET
elektronska tomografija
vzorec med zajemom mikrografij nagibamo - po 1-2° na mikrografijo, gremo od kota -60° do +60°
ločljivost je nižja kot pri SPA, večja nevarnost poškodbe vzorca (ves čas sevamo z elektroni vanj)
primerno za velike komplekse (jedrna pora)
mikro-ED
elektronska difrakcija na mikro/nanokristalih
s krio-elektronskim mikroskopom
lahko zelo visoke ločljivosti, ampak težave s poškodbami vzorca
SAXS
ozkokotno sipanje rentgenske svetlobe
isto kot XRC svetimo z X-žarki ampak molekula je v raztopini in ne kristal → vzorec sipanja izotropen
manj kontrasten, ločljivost je slabša, ampak omogoča karakterizacijo strukturno neurejenih molekul
oblika krivulje odvisna od oblike molekule (bolj majhni in kompaktni/ zviti imajo višje krivuljo)
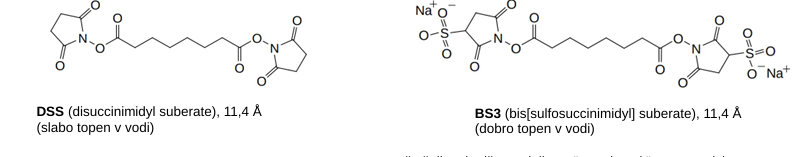
XL-MS
dobimo podatke o oddaljenosti ostankov v kompleksu
s prečnim povezovalcem povežemo random ostanke → proteoliza → dobimo različne delčke polipeptidne verige - eni so povezani skupi → masna spektroskopija
prečni povezovalci: NHS estri ponavadi (DSS in BS3)
uporabnost: modeliranje kompleksov, konformacij, ki jih zavzame protein