telomeres & transgenesis and genome editing
1/34
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
35 Terms
function of telomeres
-solve end protection problem- distinguish normal chromosome ends from ends produced by breakage
w/o would be processed in same way as ends produced by breakage- activate DNA damage response leading to p53-dependent cell cycle arrest & subsequent replicative senescence
recognition of chromosome ends by DNA repair mechanisms, e.g. non-homologous end-joining leading to chromosome fusions
-allow organisms to circumvent ‘end replication problem’ - the loss of DNA from the ends of linear chromosomes each time they replicate (linear chromosomes should shorten each time they replicate)
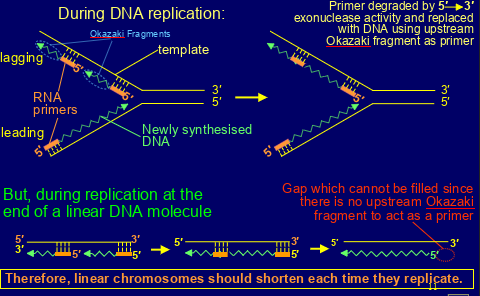
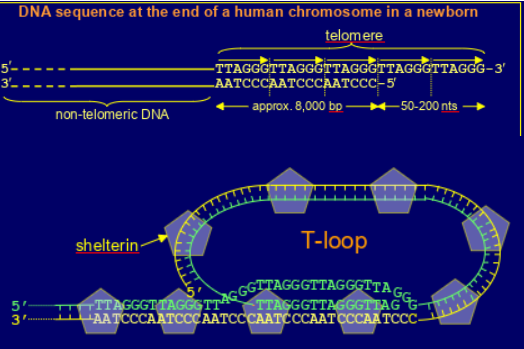
structure of telomeres
made up of tandem repeats (at birth about 8000bp) ot TTAGGG
has a short extension at the 3’ end (approx 50-200nt)
forms T-loop , bound by shelterin (complex of 6 proteins e.g. TRF1, TRF2, POT1)- prevents binding of Ku (stops triggering of DNA damage response through ATM activation) & not recognised by DNA repair systems (e.g. by Ku protein) & inhibits ATR kinase
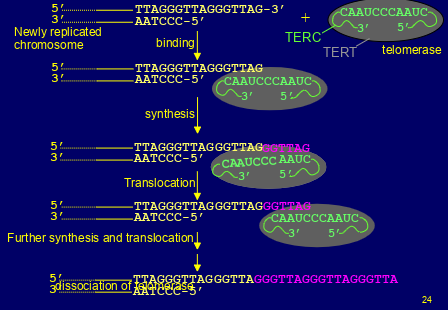
synthesis of telomeres
-maintained in germline & embryonic cells by the action of telomerase- it adds copie of the telomere repeat sequence to 3’ overhangs of newly replicated chromosomes
-telomerase absent from normal differentiated somatic cells but is in low levels in adults stem cells
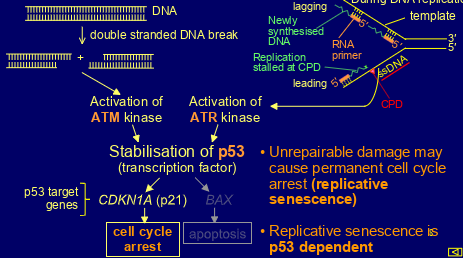
what triggers the DNA damage response
-dsDNA breaks
-presence of ssDNA
describe the DNA damage response
-dsDNA break -> activation of ATM kinase
-presence of ssDNA -> activation of ATR kinase
both kinases stabilise p53 (transcription factor)
CDKN1A (p21) then activated leading to cell cycle arrest
BAX then activated leading to apoptosis
(if damage unrepairable then may cause permanent cell cycle arrest= replicative senescence)
how much do telomeres shorten every cell division
40bp
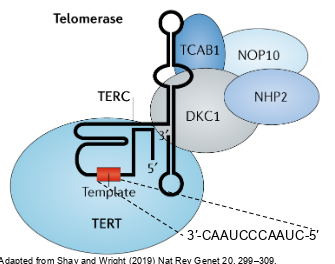
whats the structure of telomerase
-complex of RNA molecule and several proteins
-the main protein is TERT (telomerase reverse transcriptase) which is a reverse transcriptase
-the RNA molecule is TERC (telomerase RNA component) -has the seq 5’- CUAACCCUAAC-3’ which is complementary to the telomere repeat 5’-TTAGGG-3’
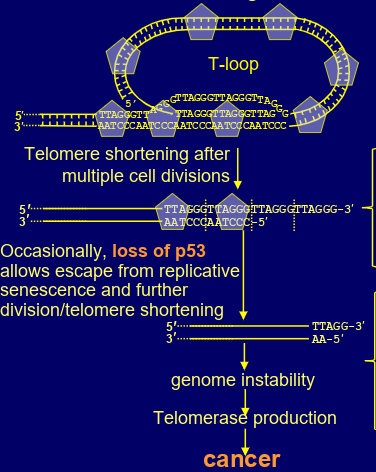
why does telomere shortening not lead to loss of genes
when telomere not long enough to make T-loop it resembles broken ends so DDR activation & replicative senescence (anti-cancer mechanism) so cell division ceases, remaining sheltering prevents operation of DNA repair mechanisms
how could telomeres induce cancer
occasional loss of p53 can allow escape from replicative senescence & further division/ telomere shortening
eventually too much telomere lost that sheltering can’t bind
DNA repair mechanisms act on chromosome ends to generate chromosome fusions
(fusion of sister chromatids ends by NHEJ then random break in mitotic spindle fibres) genome instability-> possibly leads to oncogenic mutations
tiny number of cells may activate telomerase production= become immortal cancer cells (even though almost all cells die)
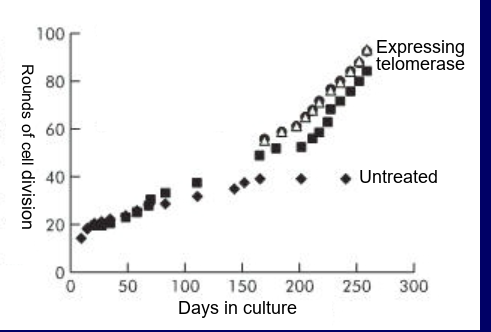
how are telomers/ telomerase related to aging?
telomerase expression makes cells immortal (keep undergoing division) (introduce TERT gene into human oesophagus cells made them immortal)
aging is correlated with shortening of telomeres
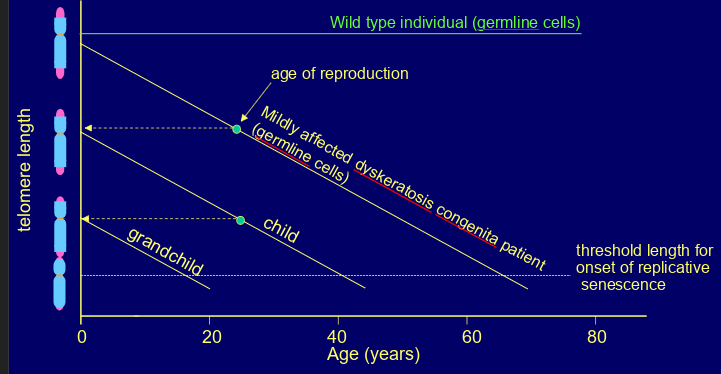
mutations in genes encoding components of telomerase (e.g. TERC or TERT) or shelterin, leading to telomeres shrinking faster than usual & shortening even in germline cells
what disorders are caused by this
dyskeratosis congenita- symptoms such as bone marrow failure, pulmonary fibrosis, liver cirrhosis, increased risk of some cancers
shows anticipation (gets worse in each successive generation) as chromosomes become progressively shorter
believed to arise from replicative senescence of stem cells(e.g. in bone marrow) leading to organ failure)
what recent breakthrough allowed more precise measures of telomere length
Nanopore & PacBio sequencing- showed telomere length varies between different chromosomes , the short and long arms of the same chromosome, the maternal and paternal copies of a chromosome
overall showed telomere length doesn’t give the whole story -some chromosomes may be much shorter than average
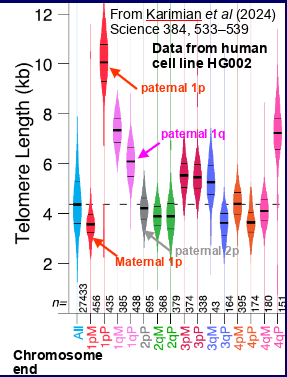
what do we not yet know about telomeres
-3d structure of a telomere
-what the threshold length of telomere DNA is below which the t-loop cant form
-how many telomeres must be lost to trigger replicative senescence
how and why would you conduct targeted integration of DNA into the S.cerevisiae genome
to study the normal function of genes in a simple eukaryote, via deleting the gene of interest from the genome and replacing w a marker gene- chemical permeabilsation of cells (treat cells with lithium acetate, polyethylene glycol, dimethyl sulfoxide, 42deg heat shock), homologous recombination-mediated integration of a selectable DNA fragment
-Homologous recombination facilitates SITE SPECIFIC integration
-Marker genes are used to identify successful recombinants
-Two-step strategies can be employed, with selection for and against markers
-Transgenesis methods allow integration of ANYTHING into the genome (is still limited e.g. deletion of essential genes cant be recovered in haploids, DNA encoding toxic product tricky(
-PCR and sequencing (or similar molecular methods) should be used to verify the insertion is correct
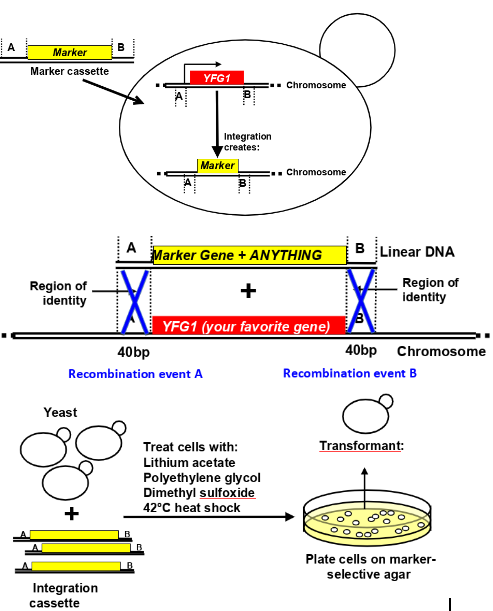
how do marker genes work
can only select for the marker gene

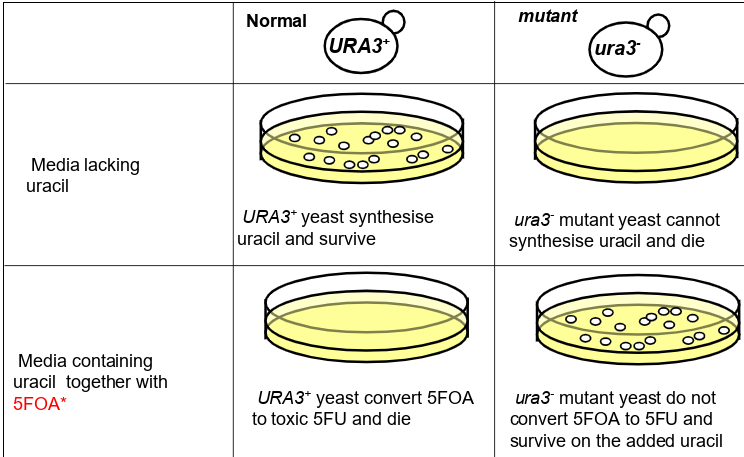
how do counter-selectable markers
can actively select for or for a different genotype: for example
You can ACTIVELY select for growth of the URA3+ genotype and death of the ura3- genotype.
You can also ACTIVELY select for growth of the ura3- genotype and death of the URA3+ genotype.
only URA3+ sythesise uracil so can be used to select against ura3-
only ura3- don’t cinvert 5FOA to 5FU that is toxic to cells do 5FOA can select against URA3+
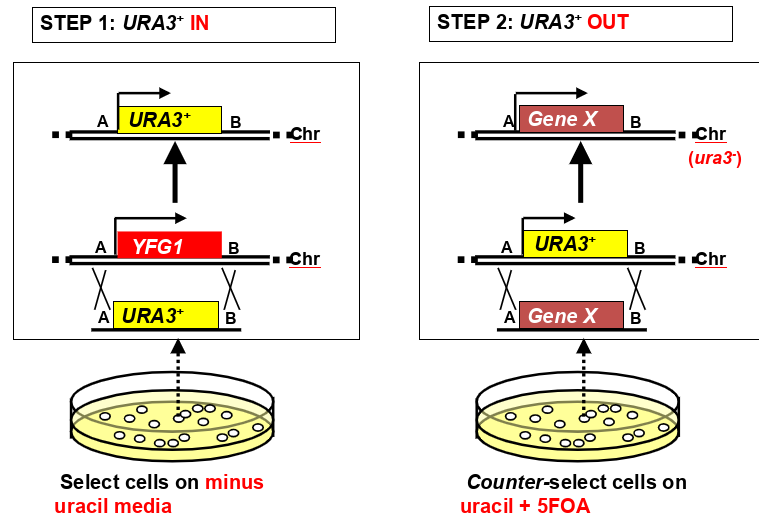
how can counter-selectable markers be used for 2-step genome modification strategies (knock in, knock out)
plate cells on minus uracil media to select for URA 3+ yfg1- integrants
plate cells on uracil and 5FOA media to counter select for ura3- integrants (replacing URA3+ gene with gene X) (gene x can be anything- deletion, site directed change, heterologous promoter, fusion protein tag, etc)
how might you identify promoter/ enhancer sites that drive tissue specific expression (example is random integration of DNA into D.melanogaster genome)
-insert a reporter gene with a basal promoter, expression will rely on being activated by genomic enhancer elements
-inject plasmid DNA into embryo leading to transposon-mediated trnasformation (inject transposase helper plasmid and vestor transposon plasmid)
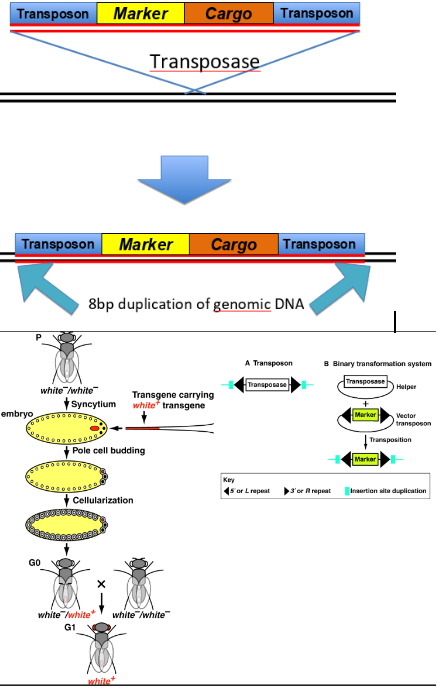
when trying to integrate random DNA into the D.melanogaster genome, what transposons could you use/ what are their features
transposons:
P-element: inserts toward 5’ end of genes, no specific target site, only works in Drosophila
piggyBac, Minos, Mariner- broad range of target species, specific target sites
(transpons can also be excised from the genome, allowing reversible transgenesis)
what marker cassettes can you use for random integration of DNA into the D.melanogaster genome
mini-white & rosy,vermillion (complements eye colour mutant)
yellow(complements body colour mutant)
3xP3-flurescent protein (expression of fluorescent fusion protein in eyes)
Adh (ethanol tolerance)
how might you express a fusion protein to label a particular tissue in the worm C. elegans (extrachromosomal arrays)
-insert multiple copies of a gene encoding GPF-fusion protein under control of a tissue specific promoter
-microinjection of DNA into worms, rearrangement of DNA to form heritable, extrachromosomal concatamers (a long continuous DNA molecule that contains multiple copies of the same DNA sequence linked in series)
describe the common marker - unc-22 antisense^b
plasmid= pPD10.46
phenotype= dominant roller
good as similar to rol-6 (dominant, easy to detect phenotype, compatible w many co-injected transgenes)
may be suppressed in some mutant backgrounds, reduced mating in males, not as easy to detect as rol-6

what key things can be learned via C/elegans DNA integration (extrachromosomal)
-Multi-copy gene arrays are silenced in the germline
-Extrachromosomal arrays don’t segregate with Mendelian properties
-Single copy insertions can be generated, it’s more uncommon
-Target sites random
-Large extrachromosomal arrays allow for large cargo fragments.
-Variable numbers of elements between lines allows for examination of both high and low level expression.
-Instability of insertions allows determination of the requirement for the gene function in different cell or tissue types.
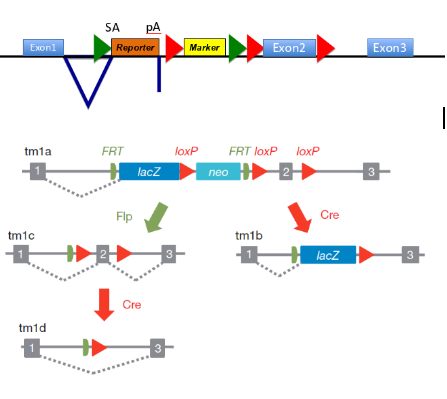
how might you generate mice with reporter and gene knockout
generate mice w one pair of recombinase sites flanking a reporter exon & another pair of flanking exons of YFG (your fav gene), deletion of region of interest in vivo
insert sites construct into embryonic stem cells via homologous recombination & generate mosaic mice → generate stable trangenic line
use site specific recombination to remove DNA flanked by either/ both of the recombinase sites
(on diagram= SA is splice acceptor site, pA is poly adenylation site- means remainder of gene not transcribed)
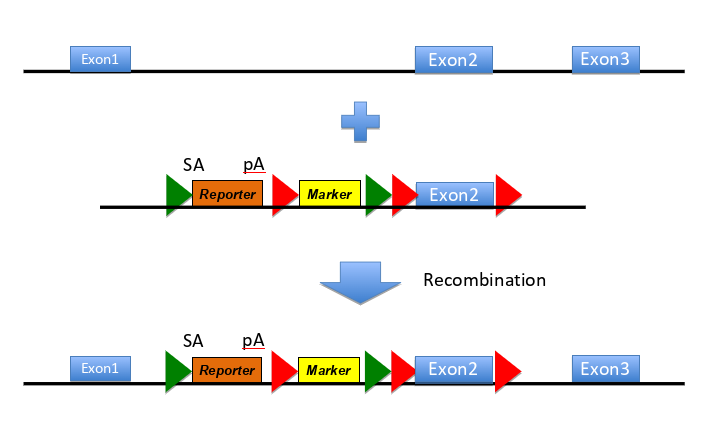
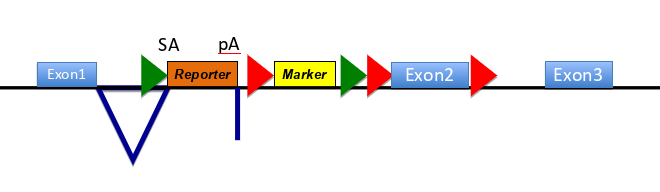
how does this construct work
-makes null allele of the targeted gene- reporter has no promoter but a slice acceptor site and polyadenylation site, meaning splicing to this exons occurs w high efficiency & the remainder of the gene is not transcribed
-reports on expression pattern of the targeted gene - reporter protein is made as an in-frame fusion w N-terminal region of the target gene (can be detected e.g. as B-galactosidase activity if reporter is lacZ)
-contains an independent marker gene to select for the integration event -marker has own promoter (red arrow) & confers resistance to a drug
-contains targets for site specific recombinases - FRT sites (green arrow) are targets for FLP recombinase so recombinase exposure deletes fragment of DNA flanked by 2 FRT sites
LoxP sites (red arrows around E2) targets for Cre recombinase, so exposure to the recombinase deletes fragment DNA flanked by 2 LoxP sites
(sequential exposure to different recombinases allows controlled deletions of 2 sections )
how can you modify the embryonic cells so they uptake the DNA more readily?
-culture ES cells from mouse pre-implantation embryos
-construct targeting vector (has DNA that are homologous to target gene, allows for targeted integration of trangenes)
-transfect ES cells, cell machinery for homologous recombination allows for targeting vector to find and recombine w target gene, could also microinject
-proliferation of targeted ES cell (use of selection)
-inject blastocyst in mouse, produces mosiac mice (heterozygous for the targeted allele)
what are the ethics of mouse research
3Rs- shouldn’t use more animals than needed
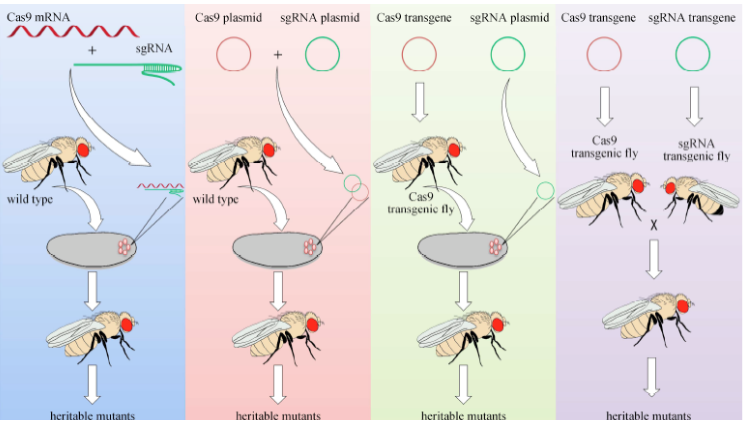
what was/ is still used before CRISPR/Cas9 for genome editing
Zinc-Finger Nucleases (ZFNs), Transcription-factor linked endo-nucleases (TALENs) -targeting achieved by sequence-specific protein-DNA interactions
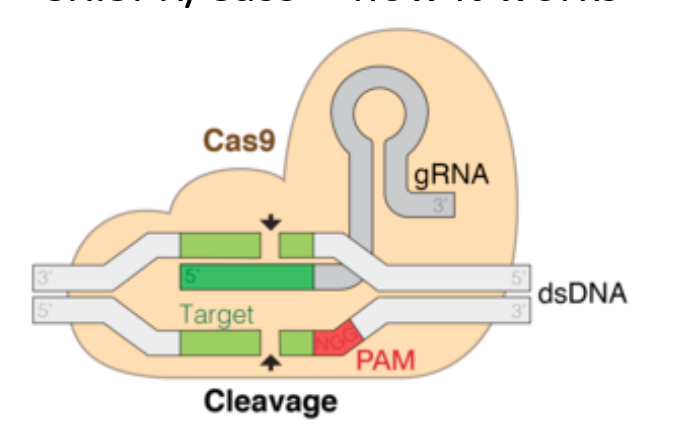
how does CRIPSR/ Cas9 work
made up of DNA cutting tool- Cas9, RNA guiding molecule- gRNA - together form Cas9 complex
locates and binds to common seq in DNA called a PAM
gRNA unwinds part of the double helix (it is designed to bind and match a particular seq in DNA) -once found correct seq, Cas9 can cut the DNA- its nuclease domains each make a nick on both side the DNA (double strand break created)
due to repair of dsDNA break error prone, inadvertently makes mutations that inactivate the gene
other enzymes can be added to nuclease domains e.g. a deaminase to convert a C-> T and therefore edit precisely genes
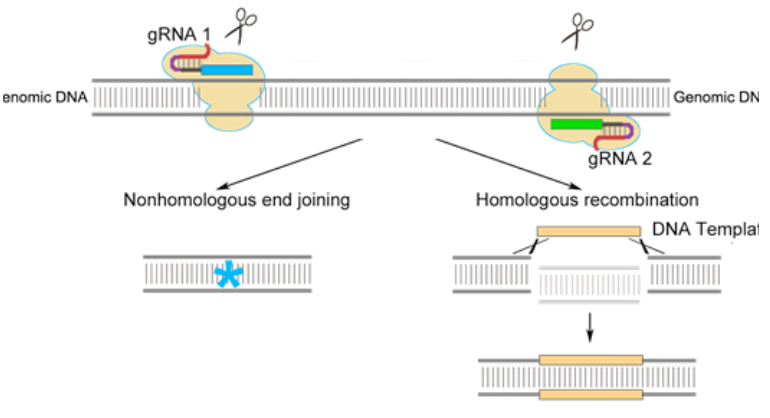
how could you utilise CRISPR to knock out a GOI in Drosophila
inject guide RNA-expressing plasmid into CAS9-expressinf flies, deletion made after repair by NHEJ, use classical genetics & molecular screening to find the mutation
how could you create a gene knock out in Drosophila (CRISPR) (again)
-inject guide RNA-expressing plasmid (need to confirm genomic target site by PCR seq before generating the gRNA construct) and donor DNA (needs to contain a selection marker & be flanked by 2 1kb homology arms) into CAS9-expressing flies
-donor DNA integrates via homologous recombination (screen for mark insertion)
-use second of recombination (CRISPR/ various methods) to delete marker & generate final strain
how can you utilise CRIPSR for producing a single point mutation
-inject guide RNA-expressing plasmid (need to confirm genomic target site by PCR seq before generating the gRNA construct) and donor DNA (needs to contain a selection marker & be flanked by 2 1kb homology arms) into CAS9-expressing flies
-donor DNA integrates via homologous recombination (screen for mark insertion)
-use PRC w primers outside the homology arms in combination w cassette primers to confirm the donor contruct in expected location
-generate and use gRNA contructs to target the region flanking the marker region
-generate and use donor plasmid w the point mutation flanked by 2 1kb homology arms containing silent (synonymous) mutations in sequences corresponding to the gRNAs
co-inject gRNAs and donor into Cas9 drosophila (and GFP+)
use genomic PCR to check for presence of point mutation
how would you generate S. cerevisiae with a loss of function in YFG1 (not allowed to use CRISPR
how would you generate C.elegans with expression of GFP in axons (no CRISPR allowed)
how would you generate C.elegans with expression of GFP in axons (CRISPR allowed)