optical microscopy
1/41
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
42 Terms
equations for light microscopy
wavelength=frequencyspeed of light energy of light=h∗f
where h is plank’s constant = 6.626×10−34 JHz−1
what is the maximum resolution for conventional light microscopy
200-300nm
what are the different ways light can interact with biological matter
absorption
reflection: specular and diffuse
scattering: elastic and inelastic
fluorescence
what is the main way contrast is produced in optical imgaging?
absorption → allows us to distinguish different biological structures producing the contrast necessary to resolve details from the background
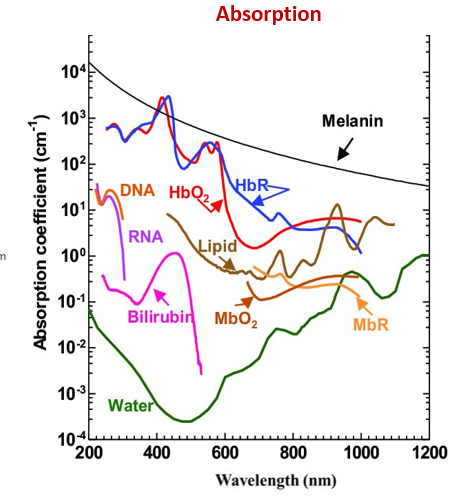
what is absorption dependent on?
the absorption coefficient of the material
the wavelength/frequency of light → high frequency/ short wavelength (blue) light is more easily absorbed
what is scattering?
types?
interaction of light photon with molecules causes a deviation from it’s original path
elastic → energy is conserved
inelastic/Raman scattering → energy is lost to the molecule which caused the interaction
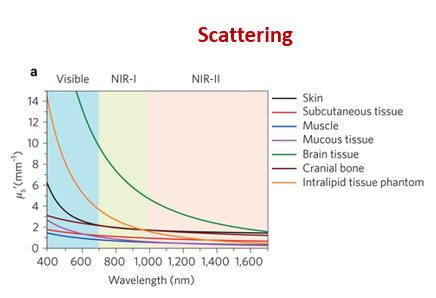
what determines noise in optical imaging?
how is it related to frequency/wavelength?
inelastic Raman scattering → determines how deep you can penetrate tissue
low frequency/short wavelength red to infrared light scatter less and can therefore penetrate deeper into the tissue
fluorescence
absorbed energy from the photon is converted into another photon type
magnification equation
magnification=actual object sizeimage size
resolution definition
smallest detail your microscope can distinguish from the background or other closely related details
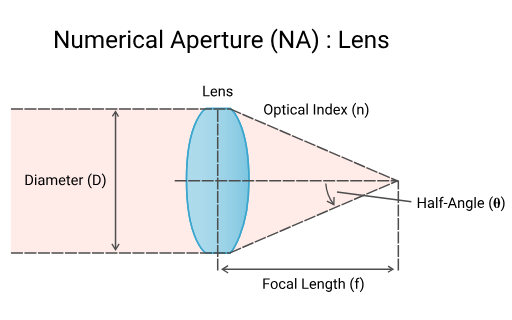
resolution equation
resolution=numerical aperture0.61 λ where numerical aperture = nsin(θ)
n: refractive index of the material between the sample and the lens ex: air = 1, oil =1.5
NA is analogous to the cone of light coming from the specimen that reaches the lens
what is the rayleigh criterion
it defines the minimum angular resolution (minimum distance at which two objects need to be, to be seen as distinct object and not a blurred blob)
when light passes through a circular aperture ex: camera lens → light isn’t focused into a perfect point → produces an airy disk
Airy disk → central bright spot surrounded by concentric faint rings
criterion states that → two points are resolved when the center of the diffraction disk of one image falls on the 1st minimum (dark ring) of the other diffraction disk
angular separation formular (no need to memorise?)
θ=1.22Dλ
D :diameter of the aperture
this equation basically tells you
the larger the aperture the better your resolution will be
the smaller your wavelength (blue or UV light) → the better your resolution → but you lose penetrative power
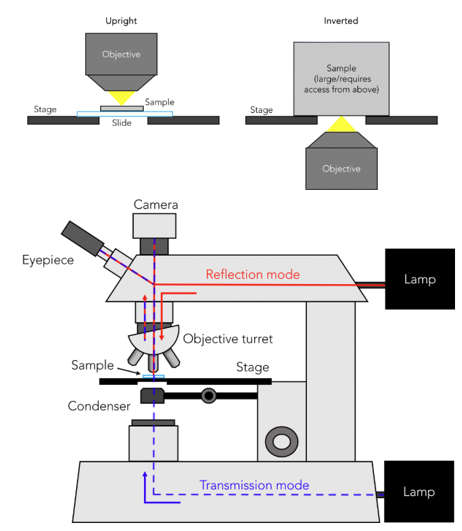
types of light microscopes
upright transmittance/transillumination microscope
fluorescence microscopy
epifluorescence microscopy
confocal microscopy
multiphoton microscopy
light sheet microscopy
super resolution microscopy
label-free microscopy
raman spectroscopy
transillumination microscope
transmittance mode:
light passes from → lamp bellow the sample → condenser → through sample → into objective lens
contrast derived from absorption of light and a bit from scattering → absorption described using beer lambert’s law
reflectance mode:
light source is located above the sample → light reflects on the surface of the sample → returns to the observer’s objective lens
used for opaque samples that does not let light through
what are the limitations of transmittance microscopy → how do we overcome them?
in thin samples: intrinsic differences in absorption are low compared with the large transmitted light from background → endogenous contrast is low
in thick samples: cells and structures are stacked on top of each other → increases scattering → increases blur
for thin samples contrast is improved using exogenous contrast agents to increase absorption ex: haematoxylin and eosin
haematoxylin: stains nuclei ribosomes and chromatin purple blue
eosin: stains cytoplasm, collagen and connective tissue pick
what is the basis of fluorescence microscopy
fluorophore absorbs a light photon → probability of absorption is described by the extinction coefficient
energy excites electrons to a higher energy level → when then fall back to stable level they produce visible light
quantum yield → ratio between number of emitted fluorescent photons vs no. of absorbed photons
lifetime: average time a fluorophore is in the excited state
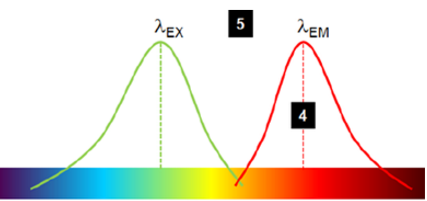
stoke shift: difference in emission and excitation wavelength → allows us to filter for the emitted wavelength → improves SNR
stoke shift
stoke shift=λemission−λexcitation
typically emission wavelength is larger than the excitation due to the loss of energy
what is multiplexing
adding different fluorophores to a single sample to distinguish different molecules and biological processes
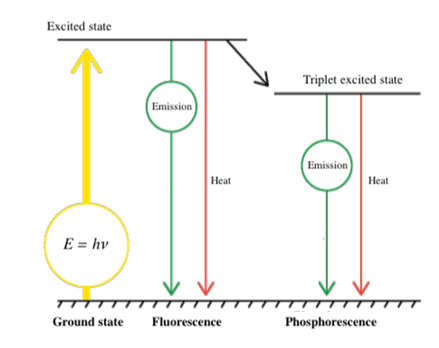
difference between fluorescence and phosphorescence
fluorescence → instantaneous production of excitation light which stops immediatly when light source is removed
phosphorescence → delayed emission of excitation light which persists after source is removed (glow in the dark effect)
how are fluorophores tagged?
fluorophores are linked to an antibody specific to a cellular protein/antigen
naturally fluorescent proteins can be genetically engineered to be produced in an animal cell ex: green fluorescence protein
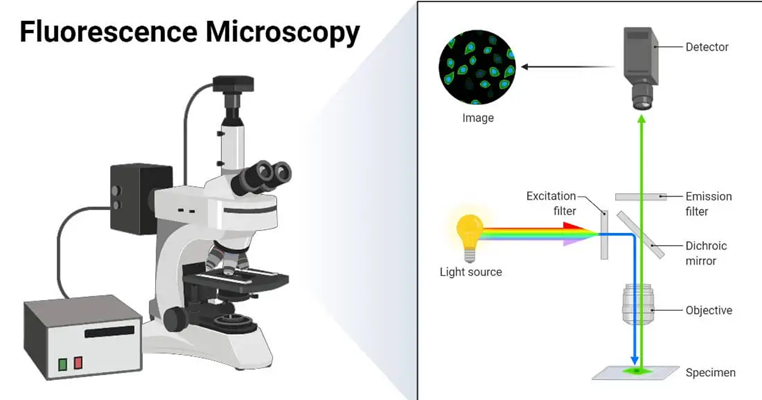
what is epi-fluorescence microscopy
light source produced above the samples if filtered → only excitation wavelength is let through
light hits sample and both excitation and emitted wavelengths are reflected back
dichroic mirror filters out emittance wavelength
produces a black background where only the emittance light makes it to the observer
limitations of fluorescence microscopy
limited resolution → 200-300
out of focus noise: some of the emitted light is scattered within the sample
when an objective focuses on a point it captures 2 cones of light → the focal plane and “wings” which are out of focus
light beam excites a central area but also some unintended areas around it due to it being a cone
produces a halo around the actual central beam which decreases SNR
this issues becomes worse with depth
photobleaching: repeated illumination of fluorophores damage them
phototoxicity: interaction between excitation light and fluorescent light can damage cells (especially short wavelength/high energy)
low number of clinically available probes
how are out of focus noise decreased?
confocal fluorescent microscopy
light sheet microscopy
multiphoton microscopy
confocal microscopy
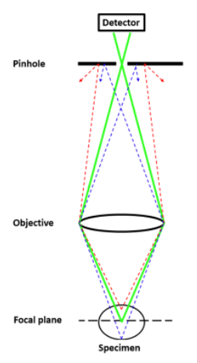
a lazer is used to decrease the volume of the light beam → reduces the cone of out of focus light → increases resolution
a pinhole is placed in front of the detector → reduces the amount of out of focus light → improves imaging depth as well
lazer is scanned across the imaging plane
stage can be moved in the z axis → samples is moved thought the focal zone → allows for optical sectioning → visualise tissue at specific depths → 3D image (confocal laser scanning microscopy) → only possible due to good out of focus rejection
increases acquisition time
depth is still limited to 500 microns due to light loss in tissue
multiphoton microscopy principles
instead of a single high energy/frequency (blue) photon → 2 or more photons at half the energy (infrared) are used to excite the sample → double the wavelength → less attenuation and scattering
if both photons hit the fluorophore at the same time their energy is summed up → emites the same photons as the single high energy photon would produce
due to non-linear absorption → multiphoton absorption is extremely rare and would only occur at the centre of the focal point due to it’s high photon density
multiphoton microscopy pros and cons
since longer wavelength/low frequency IR light is used → less scattering and absorption → increased imaging depths (1,000μm compared to 50-100μm)
large stoke shift between IR and visible light → easier filtering of excitation light from emission light
due to non-linear absorption → nearly all of emitted light must come from the focal spot centre (out of focus light suppressed) → increased spatial selectivity and sensitivity
lasers can be pulsed and has higher flux → increased probability of 2 photon absorption
cons → high acquisition time
light sheet microscopy
aims to reduce acquisition time by using a light sheet instead of a point source → produces plane by plane scanning (2D)
The laser sheet can be physical/optical (shaping the laser beam) or digital (integrating a line scanning of points in time that moves much faster than the acquisition time of the detector
this reduces photobleaching as there is a smaller irradiated area
can be used in conjunction with multiphoton excitation
disadvantage of light sheet microscopy
striping artifacts
sample features that scatter, absorb, or otherwise perturb the incident beam upstream result in weaker downstream illumination, visible as dark “stripes”.
Change in beam intensity the deeper it travels through the tissue
Corrected by modulating the light or post image processing
what is super resolution microscopy and what are the types
fluorescent imaging techniques that have a resolution bellow classical diffraction limits (<200-300nm)
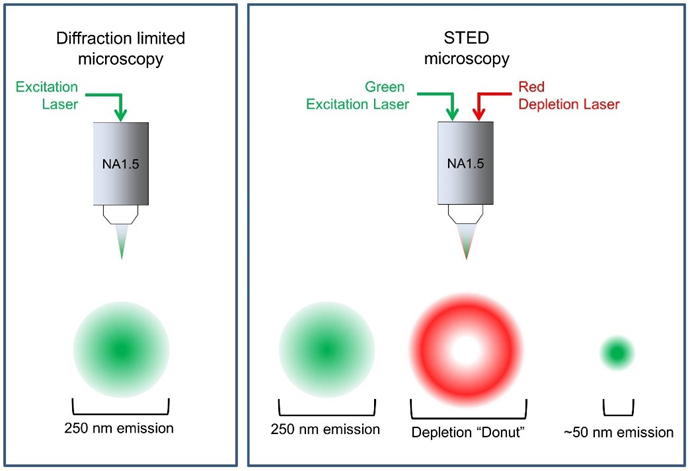
stimulated emission depletion microscopy
structured illumination microscopy
stochastic optical reconstruction microscopy
stimulated emission depletion
2 lasers are focused at the focal place:
excitation laser
depletion laser: suppresses the excited fluorophores near the excitation focal point via stimulated emission (basically photobleaches the fluorophores)
produces a donut shaped ring of suppressed fluorophores causing only a very small centre dot to be fluorescent
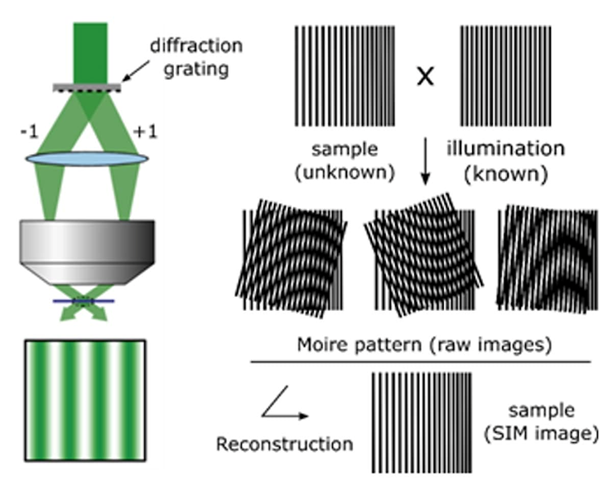
structured illumination microscopy
diffraction grating is used to illuminate sample (unknown pattern) with a known light pattern → patterns overlap
overlapping of patterns produces an interference pattern (beat pattern) called moire fringes
These patterns contain information about the sample's detail that would be invisible in a normal microscope
Computational method are then used to reconstruct the information from the pattern into a high-resolution image
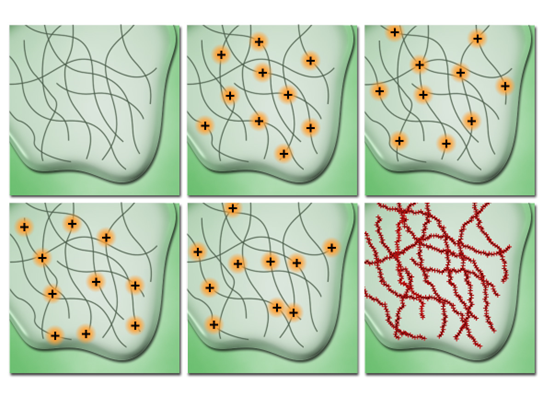
stochastic optical reconstruction microscopy
photo switchable fluorophores are used → their fluorescence can be controlled to emit a sufficient fraction of light for only a limited amount of time before bleaching to a dark state
fluorophores are excited using a laser and are trapped in a dark triplet state by a chemical buffer
a weak activation laser then illuminates the sample → only provides enough energy for a random percentage of molecules to exit dark state to glowing state
produces a stochastic rate of random fluorophores blinking
several images are taken over time → since only a few molecules are visible in each frame → system treats them as a single point → increases resolution (20nm)
what is label free microscopy + what are the types
instead of just tracking absorption we track chromophores → they absorb light differently and different wavelengths (intrinsic optical signal) → this change can be tracked to infer molecular composition or physiology
multispectral and hyperspectral imaging
raman imaging
multispectral imaging
multiple images are acquired using different continuous wavelengths of light across the whole electromagnetic spectrum
captures the difference in chromophore light absorption at that wavelength
assembles data into a 3D hypercube → x,y axis denotes positional information + z axis denotes wavelength absorption spectrum
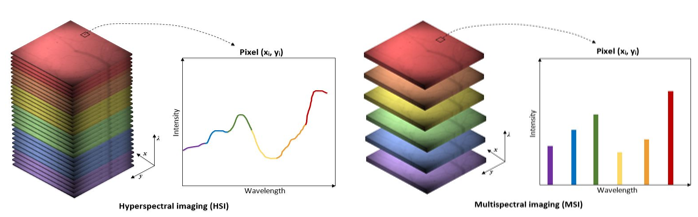
hyperspectral imaging pros and cons
pros:
quantitative information on the chemical composition of chromophores
cons:
heavy computational loads → more wavelength + more pixels
requires complex modelling of light pathlengths into tissue for quantification
poor depth resolution as it is very susceptible to the effects of scattering on spatial resolution → superficial layers or thin samples only
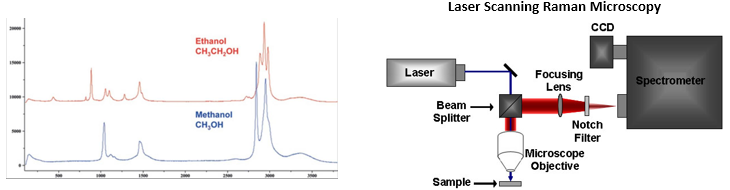
raman spectroscopy
detects inelastic raman scattering → incident light hits molecule and deviates from its path → some of the energy is absorbed by the molecules → produces a raman scattered wave with lower energy
since scattered wavelength has longer wavelength than original it produces a similar effect to the stoke shift
however this interaction has a low occurence therefore → high-intensity source (lasers) and long acquisition times (minutes to hours) are needed
the wavelength of the scattered light is dependent on chemical structure of the sample → each peak corresponds to a specific molecular bond vibration
why is tissue clearing important
scattering is the main limiting factor for depth resolution → mainly caused by lipids in cellular membrane → needs to be removed whilst preserving fluorophores and cellular matrices
need to match tissue refractive index with that of the suspending medium
what are the types of tissue clearing techniques
organic solvent method
hydrophilic method
hydrogel-based methods
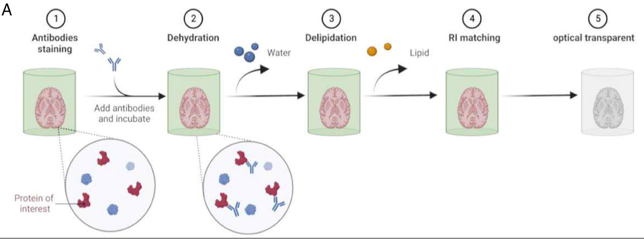
organic solvent tissue clearing
sample stained with antibody
sample completely dehydrated → causes sample to shrink (may destroy certain structures)
lipids removed using organic solvents
sample immersed in refractive index matching organic solvent
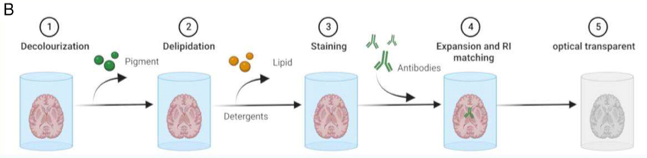
hydrophilic tissue clearing method
sample is decolourised
detergent is used to remove lipids
samples is then stained
placed in RI matching water soluble reagent
no dehydration step but can cause tissue enlargement
hydrogel based tissue clearing method
synthetic gel is made from monomers and polyepoxide → acts as a structural matrix to support tissue after delipidation
delipidation performed using detergents
sample is stained
sample placed in RI matching solution
requires longer and more complex preparation process