Pediatrics Rheumatology
1/46
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
47 Terms
Triggers of the Immune Response
Genetic Predisposition
Many genetic factors currently being studied
Infection
Reiter’s syndrome
Rheumatic fever
Lyme
Virus
Toxic
Adverse drug reaction
Environmental
Smoking?
Trauma
After tissue injury normal inflammation response fails to regulate and cascades
Unknown
Stress?
Usually have inciting incident that triggers
Manifestations of Inflammation
Synovitis
Inflammation of the joint synovium
Enthesopathy
Inflammation at the insertion of a ligament to a bone
Serositis
Inflammation of serosal lining tissue (pleura, pericardium etc.)
Myositis
Inflammation of muscle tissue
Vasculitis
Inflammation of connective tissue/vascular endothelium
Lab Testing Used in Diagnosis of Joint Pain
Tests are used as an adjunct to diagnosis and to rule out other diseases (ie. infection)
Negative testing does not always rule out a disease
May rely solely on H&P
Biopsy of tissue may be necessary for accurate diagnosis
Ie. Vasculitides such as Wegener’s, scleroderma, sarcoid
Rheumatoid Factor
Antibodies directed against immunoglobulin G
Non-specific test
Often positive in systemic lupus erythematosus (SLE), Henoch-Schonlein pupura (HSP), sarcoid
Usually NEGATIVE in Juvenile Inflammatory Arthritis (JIA)
Anti-Nuclear Antibody
Auto-antibodies against nuclear constituents in connective tissue cells
Non-specific but 60-70% of children with a positive ANA will have or will develop an autoimmune disease
Often positive in JIA, SLE, dermatomyositis
Complement Protein
Soluble proteins in the immune system
Triggers such as allergy and autoimmune disease start the complement cascade, which leads to release of tissue damaging factor
Complement may be elevated early in an inflammatory disease, along with acute phase reactants
Decreased levels of C3, C4 and total hemolytic complement C50 are seen in active SLE, SLE nephritis and other vasculidities
Histocompatibility Antigen
Human Leukocyte Antigen B27
Associated with Ankylosing spondylitis, Reiter’s Syndrome, Psoriatic arthritis, Inflammatory Bowel Disease
Human Leukocyte Antigen DRA
Associated with RF positive polyarticular JIA
Acute Phase Reactants
Various laboratory measurements which become elevated in presence of tissue inflammation
NON-SPECIFIC for any disease
Examples
Westergren erythrocyte sedimentation rate (ESR)
C-reactive protein (CRP)
Platelet count
Ferritin
Total hemolytic complement (CH50)
Other Labs to Consider
CBC
Lyme
LFT’s
BUN, creatinine
Hepatitis studies
Epstein Barr Ab
Parvovirus B19 Ab
Thyroid panel
Imaging Studies
CXR
Echo
Joint films
Bone scans
MRI
CT scan
Ultrasound
Procedures
PFT’s
Biopsy
Arthocentesis
Pericardiocentesis
JIA General
Most common rheumatologic disease in children
Biphasic peak incidence is 2-4 y/o and again at 6-12 y/o
Systemic onset equal between boys and girls
Pauciarticular (less than 4 joints affected) and polyarticular (slightly more prevalent in girls)
Etiology
Probably multifactorial
Genetic susceptibility
External triggers- trauma, infection
Pauciarticular Type 1
Younger age, peak at 2 yrs
Female predominant
Large joints are most commonly affected
Knee>ankles>elbows
Symptoms
Chronic uveitis common and may cause photophobia
Few to no systemic symptoms
Joint swelling is painless: morning stiffness and will be limping
Localized growth disturbance may occur if unilateral joint is affected
Pauciarticular Type 2
Occurs after 8 y/o
Often a strong family history of back pain, ankylosing spondylitis, IBD, Reiter’s syndrome or psoriasis
Involves joints of the lower extremity, usually asymmetrically
Knee>ankle>1st MTP
Symptoms
Early indicators include painful enthesopathy of achilles tendon, patellar tendon and plantar fascia
Acute uveitis may occur
Patients have slowly diminishing motor performance and later in disease have joint swelling
Occasional systemic signs
Fever, weight loss, anorexia, diffuse arthralgia, myalgia
JIA Lab Studies
Type I
Rheumatoid factor RARELY present
ANA positive in >50%
CBC and ESR usually normal
Type 2
Rheumatoid factor and ANA usually NEGATIVE
Children w/o constitutional symptoms have normal labs
Children with constitutional symptoms may have
Elevated ESR
Low hemoglobin
Positive HLA B27
JIA Other Studies
Imaging
X-rays initially normal: later may show joint destruction and deformities
MRI – may show acute synovial inflammation and loss of synovium
Other studies
May need arthocentesis, echocardiogram and/or synovial biopsy to rule OUT other diseases
Polyarticular JIA
Most are seronegative for rheumatoid factor
More common in girls
Involves small joints of the hands and feet, as well as larger joints
Systemic manifestations more common
Fever anorexia, fatigue
Extra-articular symptoms may include a linear salmon pink quickly fading macular rash associated with fever, lymphadenopathy, hepatosplenomegaly
Imaging may show erosive destruction of joints on x-ray
Seronegative Polyarticular JIA
RF (-)
Age usually 10 y/o or younger
Symptoms
25% have positive ANA and uveitis
Less systemic symptoms
More favorable outcome
Lab studies may show
Mild anemia
Leukocytosis
Increased ESR
Respond better to treatment with NSAIDS
Seropositive Polyarticular JIA
RF (+)
Peak age is 9-16 y/o
Symptoms
More severe disease with extra-articular symptoms
Erosive arthritis
Rheumatoid nodules develop over tendons
Labs
50% of pts have positive ANA
Other labs include increased ESR, CRP, leukocytosis
Usually persists into adulthood with course similar to adult onset RA
Systemic JIA
Least common type
Affects males and females equally
Age 16 y/o and younger
Symptoms
Presents with just systemic and no joint pain (joint pain might come later on)
Onset associated with high spiking fevers for weeks
Irritability, arthralgia, myalgia
Polyarticular arthritis not present early, occurs later in the disease
Extra-articular manifestations
Erythematous macular rash
Lymphadenopathy, hepatosplenomegaly
Chronic uveitis
Pericarditis, pleuritis
Rarely fatal except in cases of myocarditis or vascular coagulopathy
Labs
ANA and rheumatic factor are negative
Mild anemia of chronic disease
Increased WBC with left shift
Elevated platelet count
Extremely high ESR, CRP
Diagnostic Criteria
High fever for 2 weeks, arthritis in one or more joints for 6 weeks
Usually have had intermittent febrile illness may persist for years
JIA Treatment
Team based approach
Includes PCP, rheumatology, orthopedics, ophthalmology, nursing, PT/OT
Behavioral health for patient and family
Social work for school purposes
Non-steroidal Anti-Inflammatories
Inhibit prostaglandins
Need to watch for GI, renal issues
Steroids
Usually given as “pulses” in systemic JIA
Also can be given intra-articular
Immunosuppressants
Methotrexate and sulfasalazine
Will increase risk of other infection
Biologics- Immune Modulators
TNF blocker/Interleukin 1 antagonist
Entanercept, adalimumab, anakinra approved in JIA
May increase risk of TB, anaplastic anemia
IV Ig
SLE General
Often called the “great pretender” because it can effect almost any system in the body
4:1 female to male ratio before puberty
8:1 after puberty
Prevalence higher in Native American, Latin American, Asian, and African Americans
Peak age is around puberty and again in middle age, rare in children <8 y/o
Causes of SLE
Innate susceptibility
Due to complement levels, hormonal leves, immunoregulatory genes
Environmental stimuli
UV exposure, microbial response, drugs
Autoimmune proliferation
Hyperactive B/T cell activation
Defective immune complex clearance
Autoantibody production
Loss of “self tolerance”
Physical Manifestations of SLE
Mucocutaneous
Classic malar rash (70-80%)
Photosensitivity rash
Discoid rash and naso-oral ulcers
Raynaud’s phenomenon
Systemic
Fatigue
Malaise
Fever
Anorexia, weight loss
Ocular
Episcleritis, sicca syndrome
Other
Hepatosplenomegaly, edema, hypertension
Systemic Involvement in SLE
Renal
Nephritis, nephrosis, uremia, hypertension
Be careful with NSAID use
Cardiopulmonary
Myocarditis, endocarditis, pericarditis, pleuritis, pneumonitis
Hematologic
Thrombosis secondary to antiphospholipid Ab (cannot be put on OCPs)
Anemia
Gastrointestinal
Pancreatitis, mesenteric adenitis, serositis
Neuropsychiatric
Seizure, stroke, psychosis, peripheral neuropathy, headache, aseptic meningitis, myelitis, depression
SLE Lab Testing
ANA- positive in almost all patients
C3,C4, C50 usually low
Antiphospholipid Ab- associated with thrombosis
Anti-DS DNA, Anti-Smith Ab
CBC- leukopenia, thrombocytopenia, hemolytic anemia, reticulocytosis
Chemistries- evaluate renal function
Urinalysis- proteinuria, cellular casts
Increased ESR, CRP
Anti-histone antibody- if drug-induced lupus suspected
SLE Other Diagnostic Testing
Imaging
CXR, EKG
Renal U/S
High resolution CT to evaluate for pulmonary fibrosis
MRI of brain if neurologic involvement
Other studies
Pulmonary function tests
Renal biopsy
Tissue or skin biopsy
SLE Treatment
Dependent on manifestations
NSAIDS
Musculoskeletal pain, arthritis
Hydroxychloroquine
Skin and joint involvement
Oral or IV pulse steroids
Wide spread organ system involvement, renal disease
Cytotoxic agents
Cyclophosphamide, azathioprine, mycophenolate
Reserved for severe disease not responding to other treatment
Biologics
Monoclonal antibody- rituximab
Other supportive symptom treatment
Anticoagulants
Patients with positive anti-phospholipid antibody or thromboses
Anti-hypertensives
Patients with renal disease
Calcium/Vitamin D supplements
Patients with arthritis, or on steroids
Anti-seizure, antidepressants
Patients with neurological and psychiatric symptoms
Dialysis, kidney transplant
Henoch-Schonlein Purpura General
Most common vasculitis in childhood
IgA mediated vasculitis
More common in males
In ½ to 2/3 of children a viral URI precedes the clinical onset of HSP
Typically self limiting, but 1/3 of pts have 1 or more recurrences
Causes of HSP
Autoimmune reaction thought to be triggered by exposure to infection or environment
Infectious triggers → rhinovirus, adenovirus, EBV, mycoplasma, parvovirus B19, Gp A Strep
Vaccines
Drugs
PCN’s, quinine based
Cold exposure
Insect bites
Complications of HSP
Acute and chronic glomerulonephritis
Acute nephrotic syndrome
Intussusception and ischemic bowel
Hepatomegaly, hydrops of gallbladder
Headache, and rarely seizure, paresis, coma
Pulmonary hemorrhage secondary to vasculitis
Clinical Manifestations of HSP
Most children present following URI
Symmetrical purpuric papules and plaques on lower extremities
GI symptoms follow
Nausea, vomiting, abdominal cramping/pain, hematochezia
>60% have arthralgia and more rarely joint swelling, scalp and scrotal edema
Headache, irritability
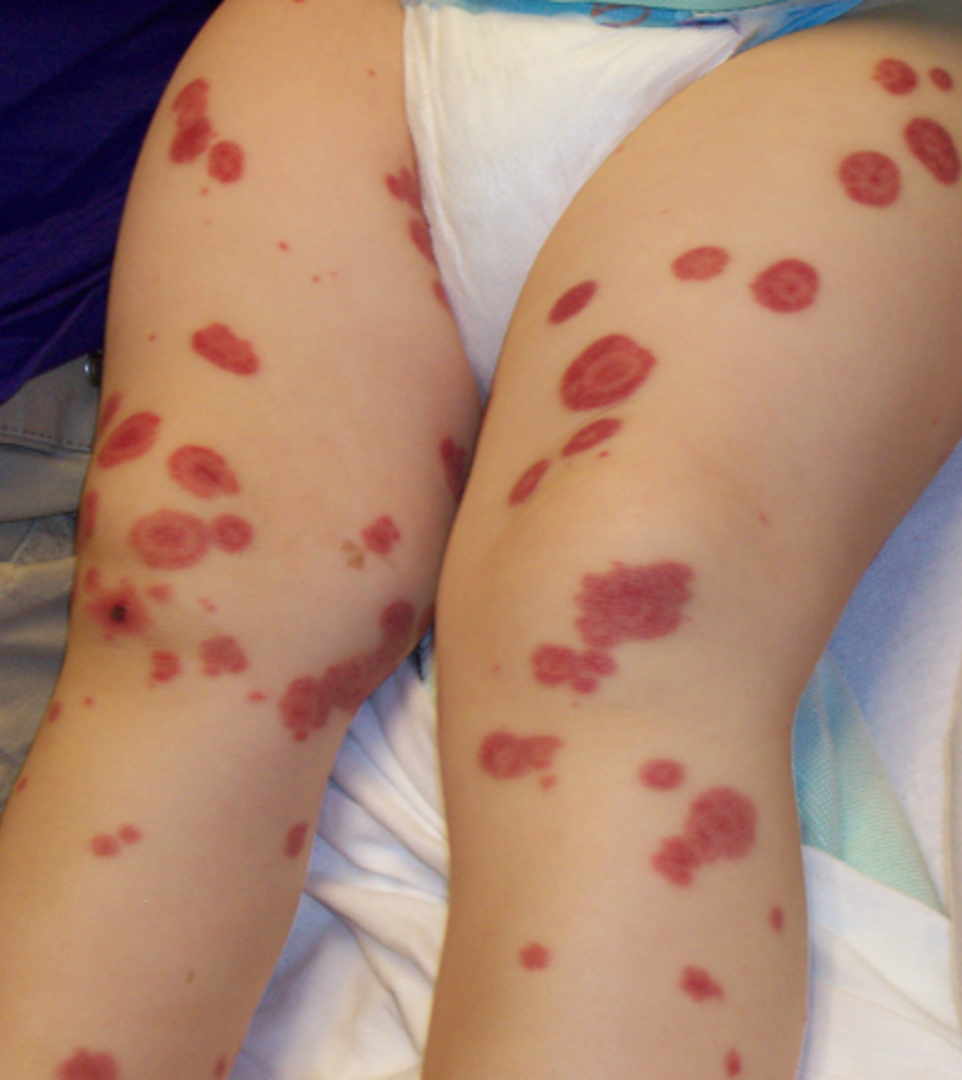
Lab Studies in HSP
CBC- leukocytosis and thrombocytosis
Urinalysis- hematuria, +/- proteinuria
ANA and RF negative
Serum IgA increased in 50% of pts
Complement levels often decreased
BUN/ creatinine elevated with renal involvement
Treatment of HSP
Most cases self limiting and only require monitoring for GI and renal complications
Nephropathy is also initially treated conservatively
Steroids as needed to relieve symptoms
Immunosuppressants have no role in HSP but may be used if chronic glomerulonephritis develops
Scleroderma General
Tissue fibrosing disorder- rare in children
Children develop a localized form, involving skin
Morphea- patch like area on trunk or head
Linear- extremities
Progressive systemic sclerosis is the systemic form
Involves all systems, especially kidneys, GI tract
CREST syndrome- milder variant of PSS
Calcinosis, Raynaud phenomenon, Esophageal hypo-motility, Sclerodactyly, Telangiectasia
Clinical Symptoms of Localized Scleroderma
Initially presents as inflamed purplish lesions with raised edges
Later progresses to hypopigmentation, skin thickening and atrophy
May involve muscles, ligaments, bone
Can lead to contractures and limb undergrowth
Morphea lesions may soften and regress
Clinical Symptoms of Progressive Scleroderma
Systemic scleroderma
Raynaud phenomenon is often first sign of disease
Involvement of digits common, causing flexion contractures
HTN due to kidney involvement
Esophageal and intestinal hypo-motility
Restrictive pulmonary disease, pulmonary hypertension
Lab Testing for Scleroderma
No specific test, clinical diagnosis
But ANA, anti-centromere, and anti scl-70 are often positive
Scleroderma Treatment
Localized and systemic scleroderma are treat with a variety of medications including
Hydroxychloroquine, corticosteroids, methotrexate and other anti-rheumatic drugs
Physical therapy often helpful
Splinting of extremities involved to prevent permanent damage
Juvenile Dermatomyositis General
Inflammatory disease involving skin and striated muscle tissue
Affects children <18 y/o
Diagnosis based on 5 criteria
Characteristic skin rash
Proximal muscle weakness
Elevated muscle enzymes
Abnormal electromyography
Abnormal muscle biopsy
Very rare (2-4 per 1,000,000)
More common in girls
Typical age presentation 7-8 y/o
Juvenile Dermatomyositis Clinical Manifestation
Heliotrope facial rash
Gottren papules on fingers
Purplish rash in sun-exposed areas
Telangiectasias in nail folds
Proximal muscle weakness
Rarely dysphagia or respiratory muscle compromise
Juvenile Dermatomyositis Diagnostic Testing
Blood tests not helpful
ANA positive in 50%
Will see elevated CK
Electromyography shows decrease proximal muscle function
Muscle biopsy shows atrophy and inflammatory cell infiltration
Juvenile Dermatomyositis Treatment
Corticosteroids
Immunosuppressants
Cyclophosphamide, methotrexate, hydroxychloroquine
IV Ig in steroid resistant patients
PT/OT
No cure, some patients go into remission, some progress
Wegener’s Granulomatosis General
Rare in children, more commonly diagnosed in teens and adults
Triad of manifestations
Necrotizing granulomas of the upper and lower respiratory tract
Necrotizing vasculitis of arteries and veins
Focal necrotizing glomerulonephritis
Think lungs and kidneys with significant hemoptysis
Wegener’s Granulomatosis Physical Manifestations
ENT
Sinusitis, subglottic stenosis, hearing loss
Pulmonary
Tracheobronchitis, tracheal and bronchial narrowing, pulmonary infiltrates, nodules, hemoptysis
Renal
Glomerulonephritis with hematuria, proteinuria
Ocular
Episcleritis, dacryocystitis
General
Arthralgia, weight loss, non-specific rash
Vascular inflammation and formation of granulomas
Wegener’s Granulomatosis Diagnostic Testing
Elevated ESR and CRP
Antineutrophil cytoplasmic antibody
Positive in 40-90% of patients with active disease
CXR/CT- may show nodules or infiltrates
Biopsy of sinus, bronchi, lung, kidney will show necrotizing granulomas and is diagnostic (very difficult and have to be very specific)
Wegener’s Granulomatosis Treatment
Corticosteroids
Immunosuppressants and immune modulators
Antibiotics for chronic infections
Surgical treatment of sinus disease and subglottic stenosis