Opioid-Derived Analgesics & Therapeutic Agents (Hale)
1/34
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
35 Terms
Endogenous opioid peptides are primarily synthesized in the central & peripheral nervous systems
Key families of endogenous opioid peptides include endorphins, enkephalins and dynorphins
Most opioid peptides share the common N-terminal sequence (H2N-Tyr-Gly-Gly-Phe-)
Endogenous opioid peptides bind to and activate (agonize) a family of G-protein coupled receptors in the spinal cord and brain to exert an analgesic effects (mu and kappa)
Endogenous opioid peptides have not been successfully developed as therapeutics largely due to their poor ADME properties
Endogenous Opioid Peptides
GPCRs
Acute receptor activation decreases neuronal excitability
- Inhibition of adenylate cyclase
- Decrease in cyclic adenosine monophosphate (cAMP)
- Modulation of K+ and Ca++ channels
- Recruitment of β-arrestin
Chronic receptor activation can lead to tolerance
-receptor desensitization from phosphorylation and decreased downstream signaling, beta-arestin recruitment and internalization
Chronic receptor activation followed by sudden cessation of agonist demonstrates dependence
-Rapid increase in cAMP levels across cells/organs is thought to be the driver of withdrawal symptoms
-Rewarding effects of receptor agonism coupled with aversive nature of withdrawal symptoms is a driver of addiction
Opioid Receptors (ORs)
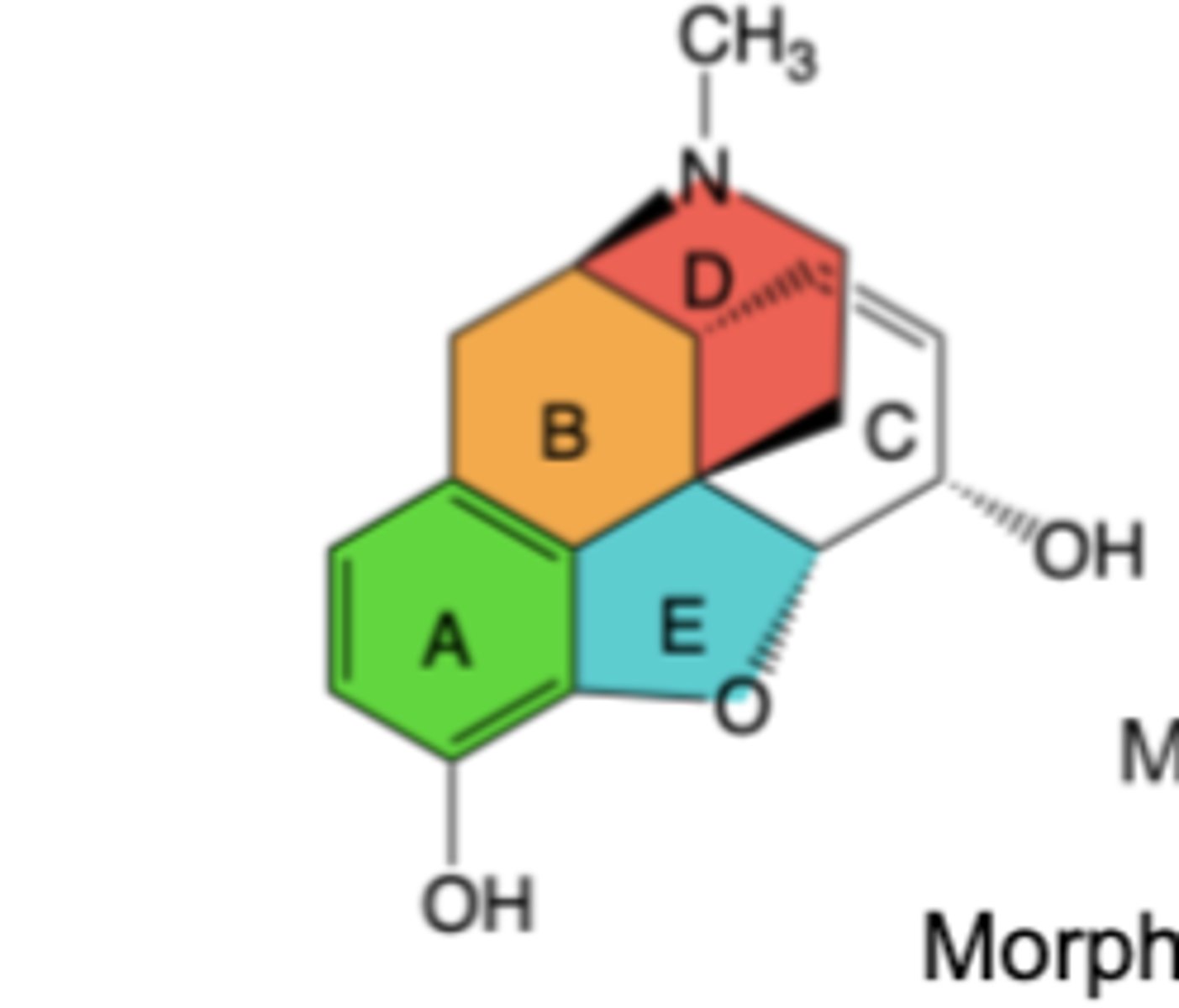
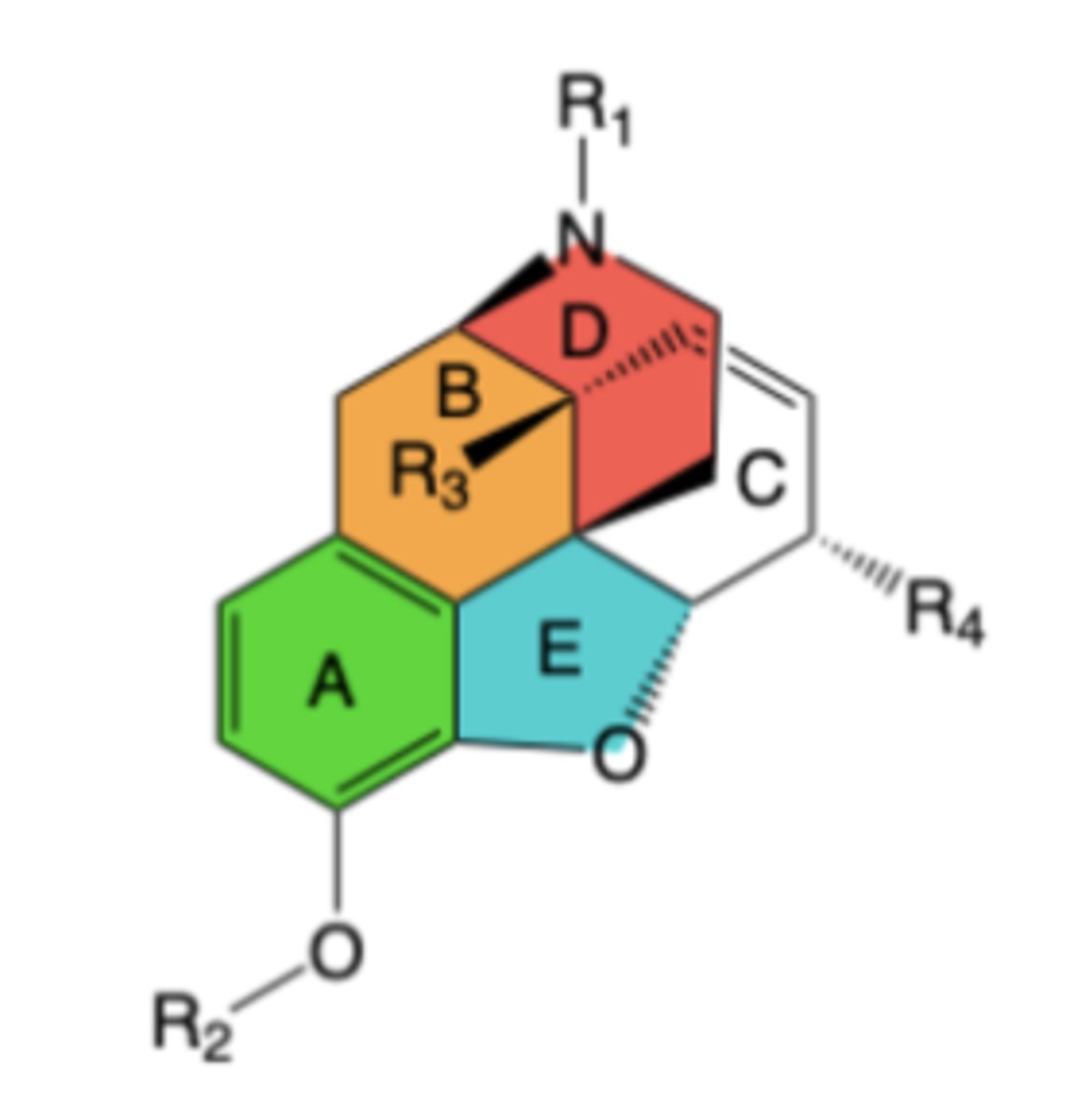
-Morphine is a selective μ-opioid receptor agonist
-Chemical structure has 5 rings and 5 chiral centers
Key chemical structure features
–A ring: Configuration efficiently mimics receptor-side chain interaction of the N-terminal tyrosine of the enkephalins
–A & D rings: Form a 4-phenylpiperidine group
B/C ring fusion: cis is preferred in morphine analogs -> trans ring fusion is 10-times less potent
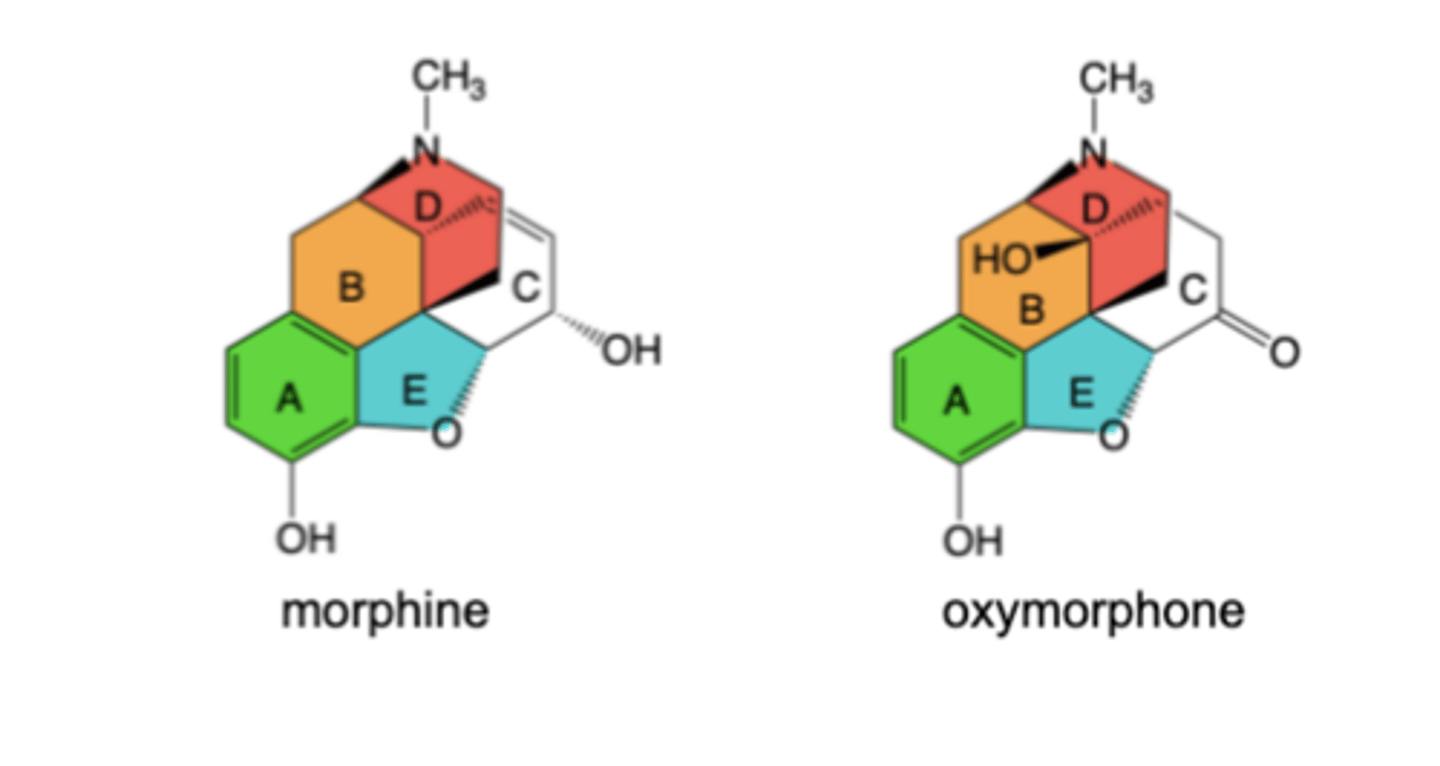
Oxymorphone is a close structural analog of morphine
-Structure differs by addition of C14 hydroxy group and isomerization of C ring (allylic alcohol -> ketone)
-Oxymorphone is a highly potent and selective -opioid receptor agonist -> 10x more potent than morphine
-Rigid chemical structure enables multiple, stereospecific interactions with mu receptors
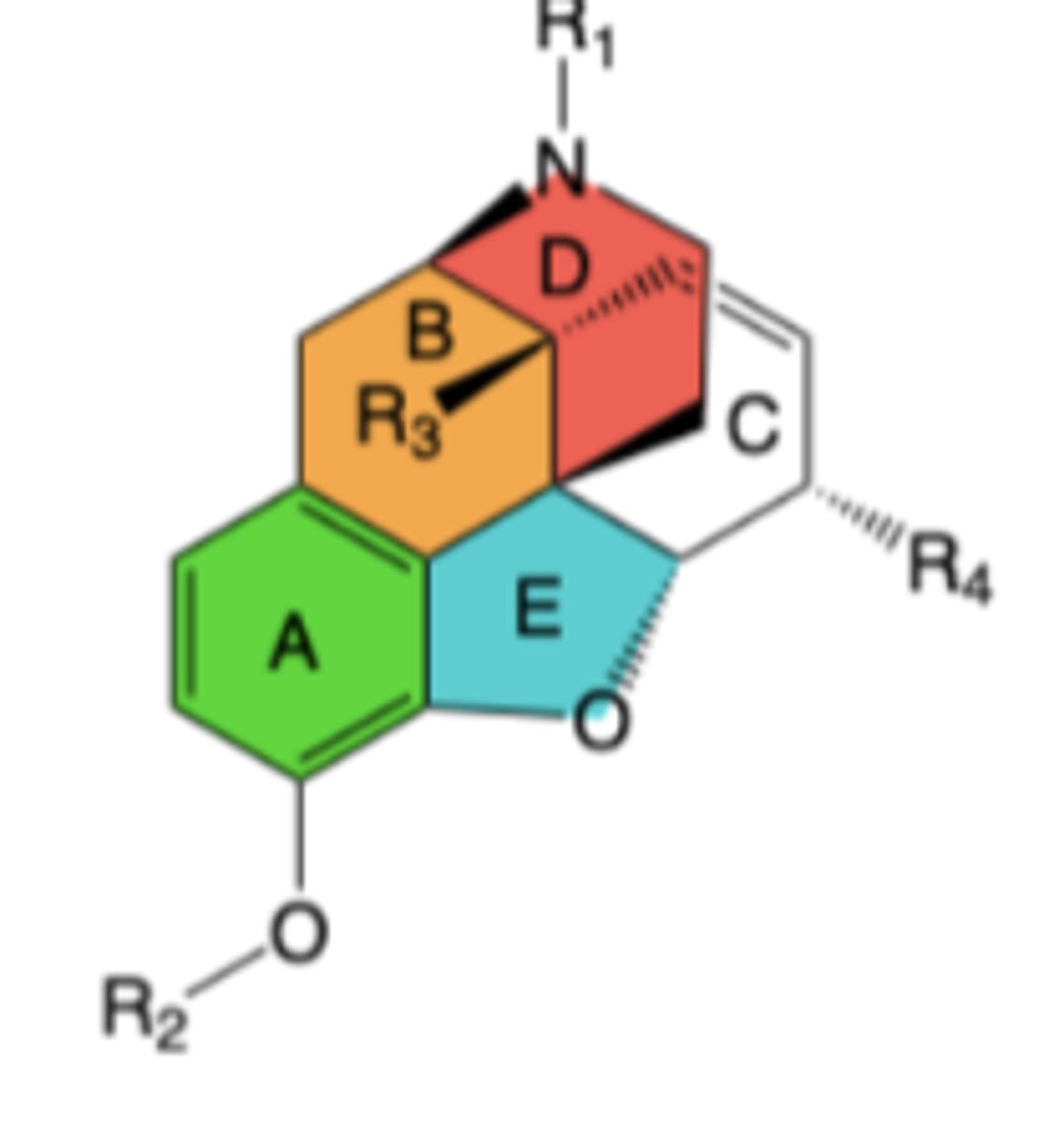
R1:
-Receptor binding: trisubstituted amine is essential for binding and to promote brain penetration;Larger alkyl groups can bind well, but diminish agonism and/or introduce antagonist activity
-Receptor agonism: R1 = methyl (-CH3) is highly preferred for agonism
R2:
-Receptor binding and agonism: R2 hydrogen (-H) is hihgly preferred for both
-R2 methyl or acetyl reduce binding but can act as prodrugs
R3:
-R3 hydrogen goof for receptor binding an agonism
-R3 hydroxy enhances binding but can decrease brain penetration
R4 and C ring:
-Receptor binding and agonism: R4 hydroxy when C ring has double bond to form allylic alcohol or R4 keto with saturated C ring are good for both
-R4 hydroxy with saturated C ring decreases binding
-R4 acetyl reduces binding but can act as prodrug
Structure-Activity Relationships (SAR) of mu-Agonist Morphine Congeners
• Morphine
• Heroin
• Codeine
• Hydromorphone
• Oxymorphone
• Oxycodone
• Hydrocodone
• Levorphanol
Morphine Congeners (Mu Agonists)
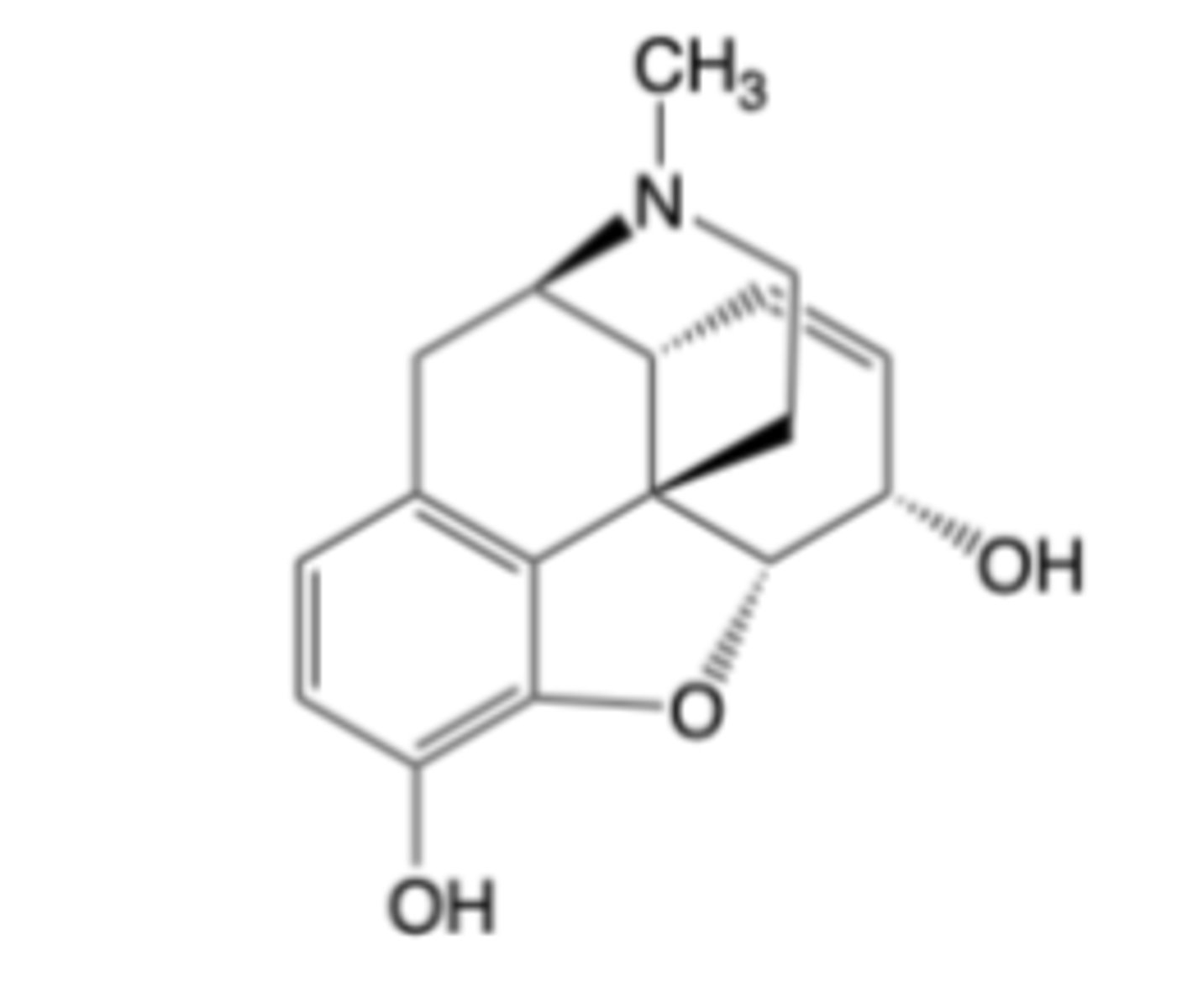
Morphine
-Indicated for the management of acute and chronic pain severe enough to require an opioid analgesic and for which alternative treatments are inadequate
-Available in formulations of sulfate salt for oral, ER, rectal, and IM
-LIMITATIONS OF USE: Due to abuse, misuse and addiction risks, morphine use is reserved for patients for whom alternate treatment options are inadequate
MOA: Potent, selective mu-opioid receptor agonist
-Active metabolite (morphine 6-glucuronide) is significant contributor to efficacy
-CI: Acute or severe bronchial asthma -> due to stimulation of histamine release
-Warnings: respiratory depression, adrenal insufficiency, severe hypotension, etc.
-AEs: Constipation, nausea, somnolence, lightheadedness, dizziness, sedation, vomiting, sweating
-DDIs: Benzodiazepines, serotonergic drugs, monoamine oxidase inhibitors, muscle relaxants, diuretics, anticholinergic drugs, P-gp inhibitors
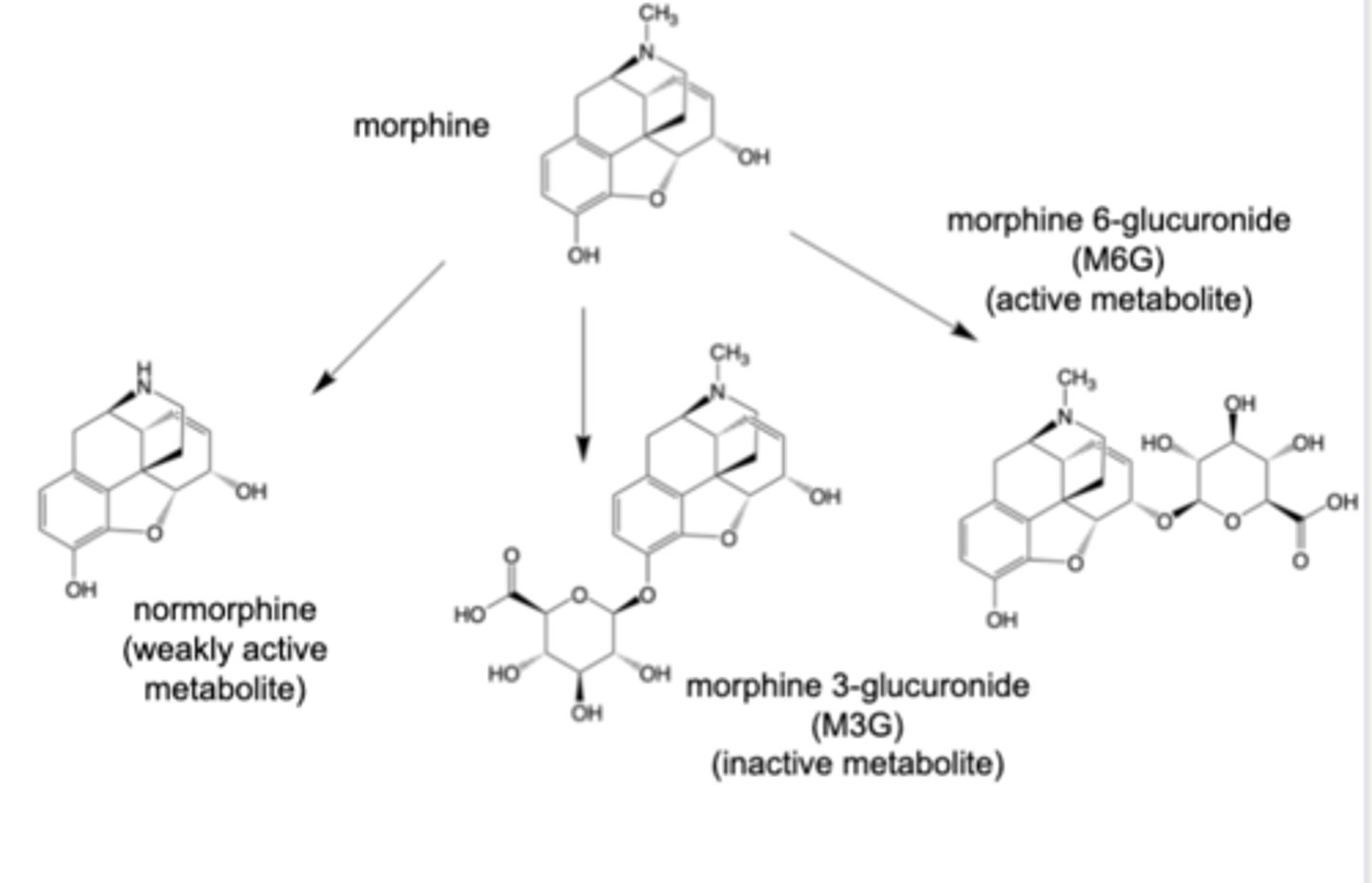
Normorphine
-Formed by CYP3A4 oxidation
Morphine 3-glucuronide (M3G)
-Formed by action of UGT2B7 enzyme
-~50% of oral dose converted M3G
-inactive, but undergoes extensive enterohepatic cycling (to morphine)
Morphine 6-glucuronide (M6G) --> main contribution to analgesic effect
-Formed by action of UGT2B7 enzyme
-5% of oral dose converted to M6G
– More potent mu agonist, but poorer at crossing the BBB (than morphine)
– Contributor to analgesic efficacy
Metabolism of morphine
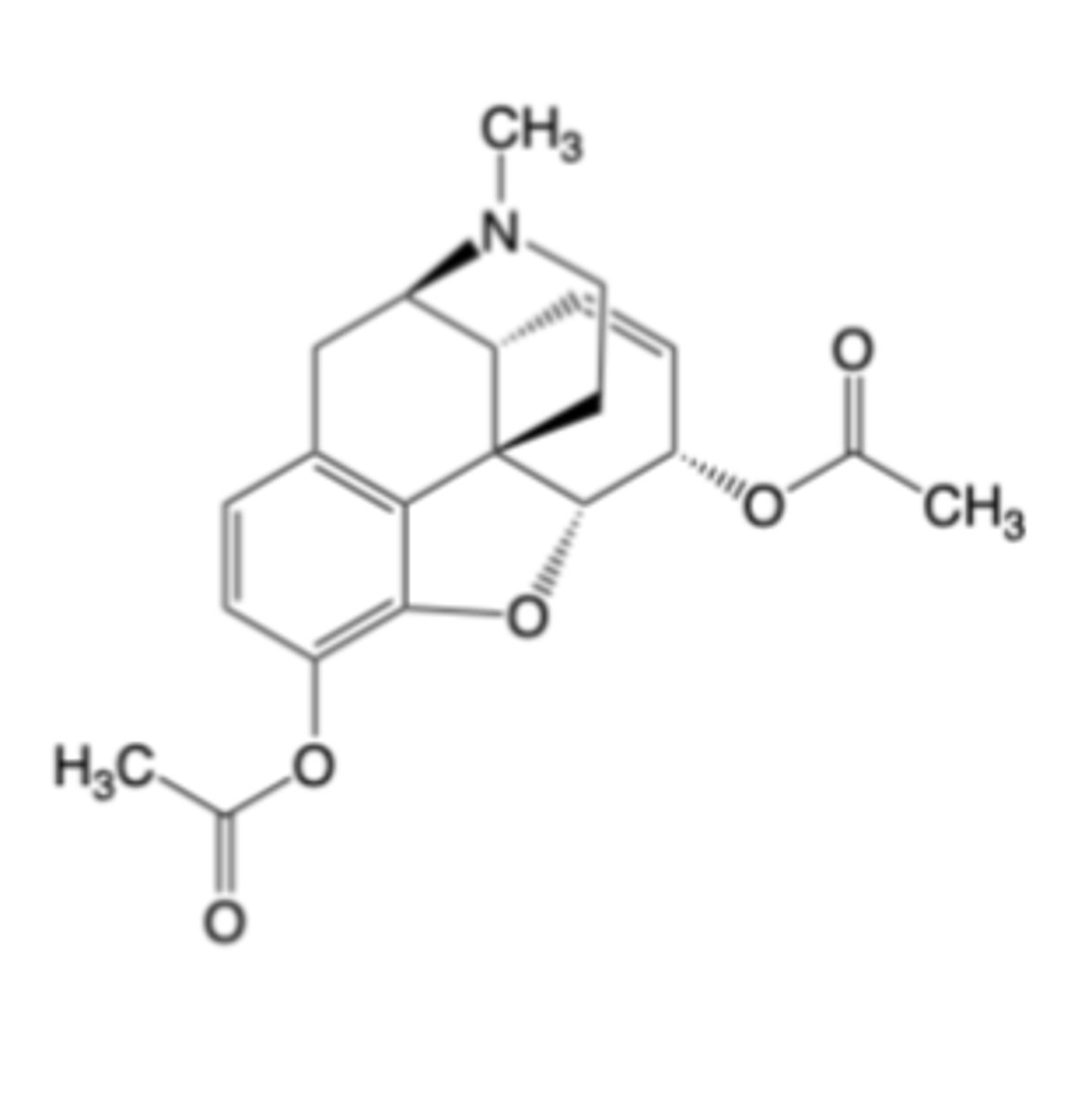
Heroin
-Initially commercialized as a cough suppressant and respiratory disease remedy
-Heroin is deacetylated by plasma & tissue esterases regardless of route of administration
-Schedule 1 controlled substance, no legitimate medical use due to severe dependence and widespread illicit use
IV heroin (to rats) delivers potent mu receptor agonists to the brain much more efficiently than does IV morphine
Hydromorphone
-Indicated for the management of acute and chronic pain severe enough to require an opioid analgesic & for which alternative treatments are inadequate
-LIMITATIONS OF USE: Due to abuse, misuse and addiction risks, hydromorphone use is reserved for patients for whom alternate treatment options are inadequate
MOA: Highly potent, selective mu-opioid receptor agonist -> ~4-7x more potent than morphine due to C ring ketone
everything similar to morphine, just more potent due to C ring ketone
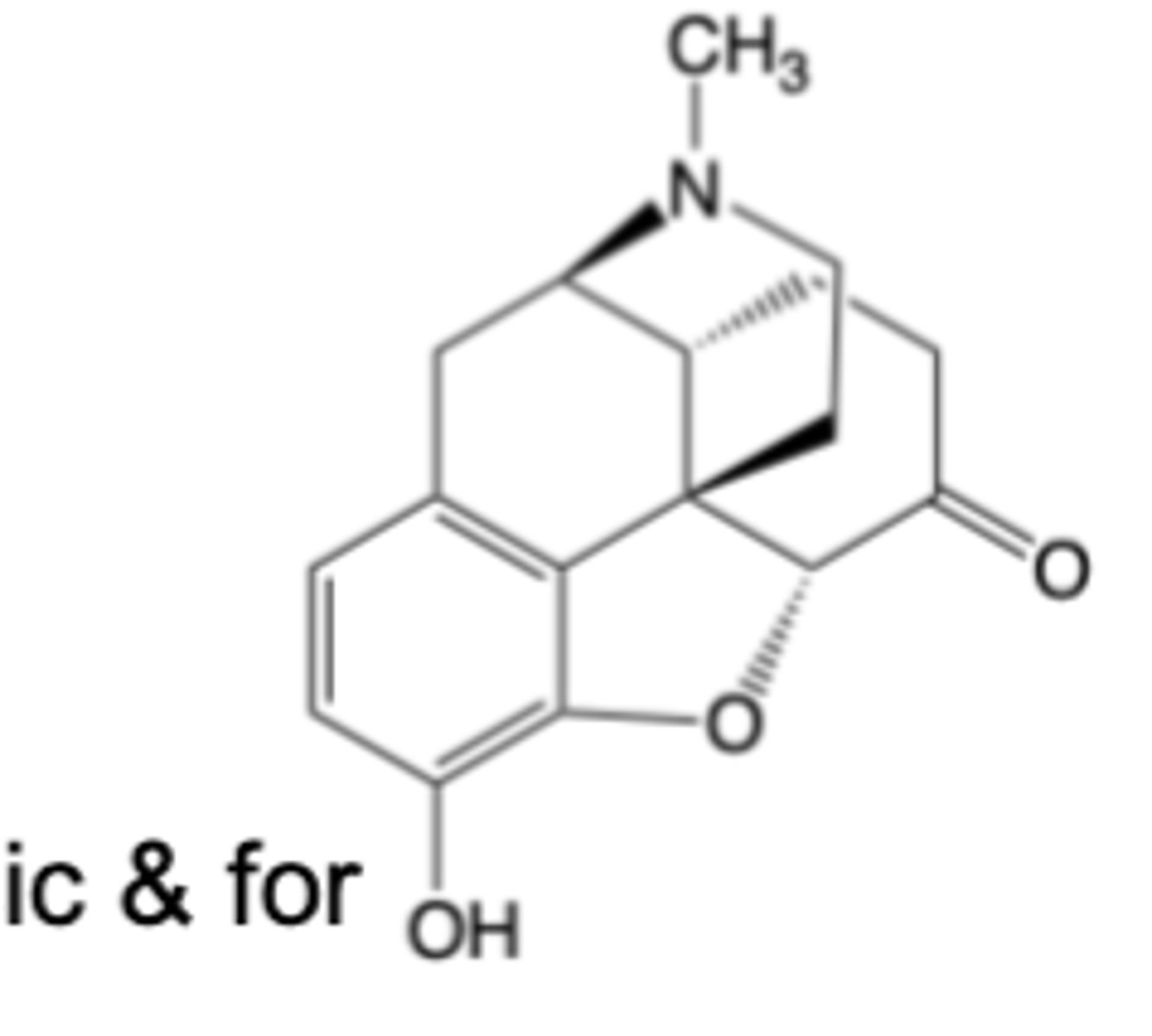
Oxymorphone
-Indicated for the relief of moderate to severe pain in patients requiring continuous around-the-clock opioid treatment for an extended period (e.g., post-surgical pain)
MOA: Highly potent, selective -opioid receptor agonist -> 10x more potent than morphine due to C ring ketone + 4alpha-OH
Longer t1/2
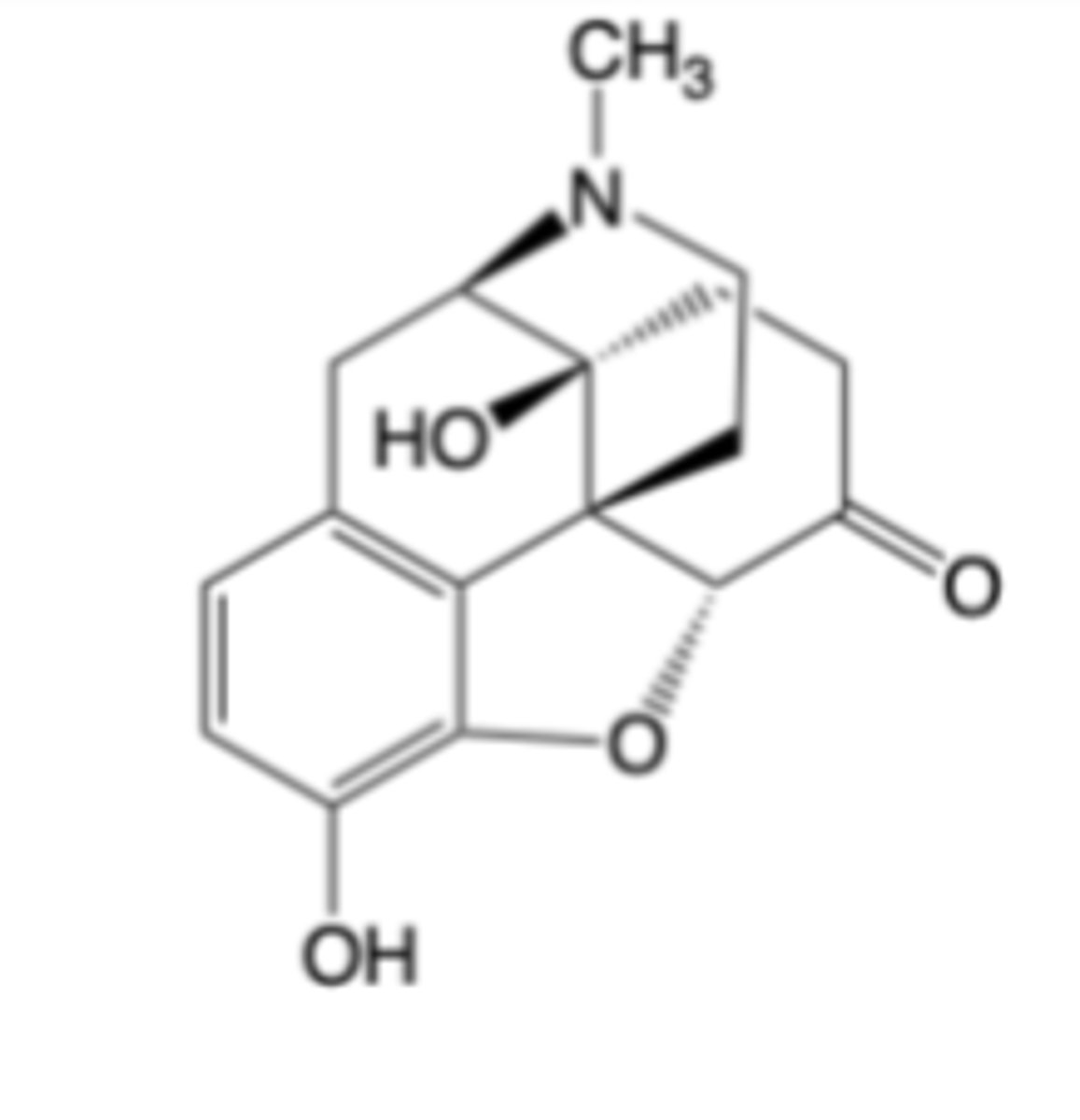
Codeine
-Indicated for the management of mild to moderate pain where treatment with an opioid is appropriate & for which alternative treatments are inadequate
-More frequently co- formulated with non-opioid analgesics or with an antihistamine, expectorant and/or decongestant for cough (both ex-U.S.)
-single oral formulation (higher BA than morphine)
MOA: Weakly potent, selective mu-opioid receptor agonist -> ~10x less potent than morphine
Dose for antitussive (cough suppressant) activity is generally lower than that needed for analgesia
CI: Contraindicated for use in children less than 12 years old (due to CYP 2D6 polymorphism), Acute or severe bronchial asthma (due to stimulation of histamine release), MAOIs, GI obstruction
Primary AE: Constipation
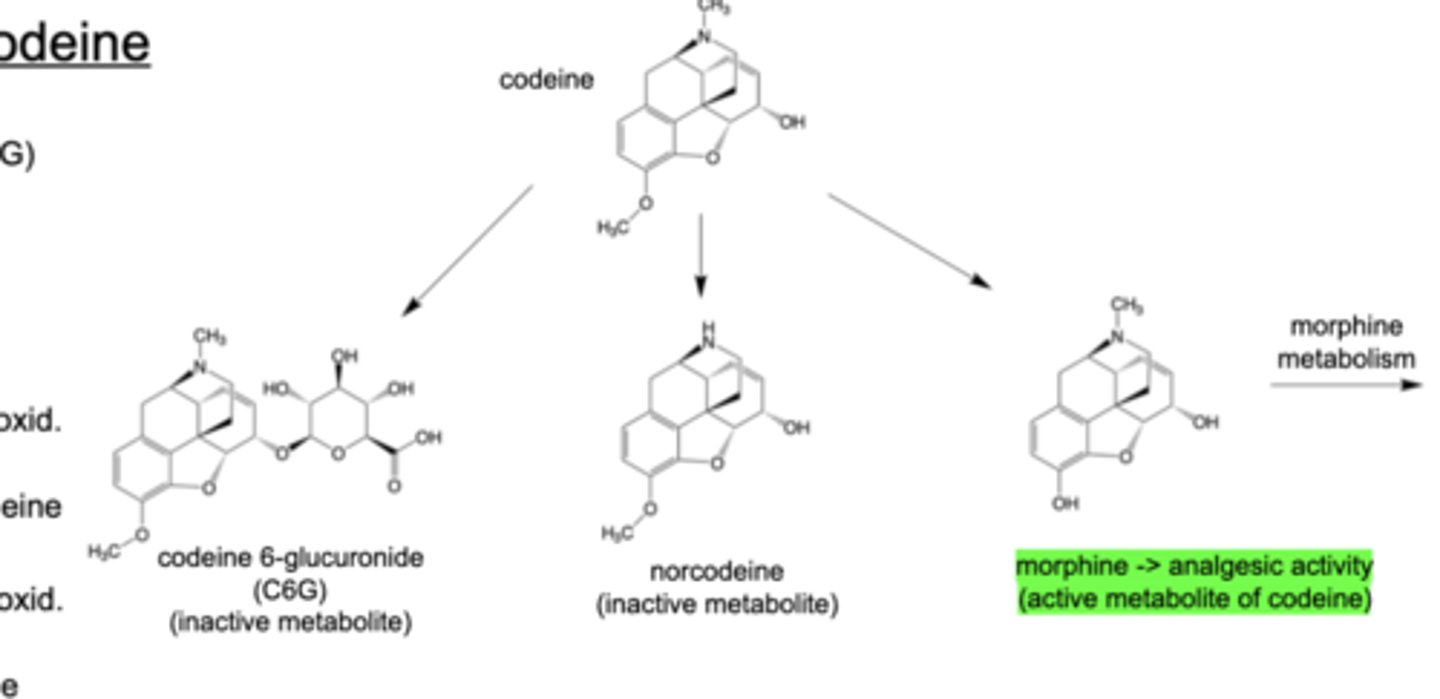
Codeine 6-glucuronide (C6G)
-Formed by action of UGT2B7 enzyme (50-70% of oral dose)
Norcodeine
-Formed by CYP3A4 oxid. (10-15%)
Morphine
-active metabolite of codeine --> analgesic activity
-Formed by CYP2D6 oxidation (5-15% of oral dose converted to morphine)
-Life-threatening respiratory depression and death have occurred in children due to a CYP2D6 polymorphism
Metabolism of Codeine
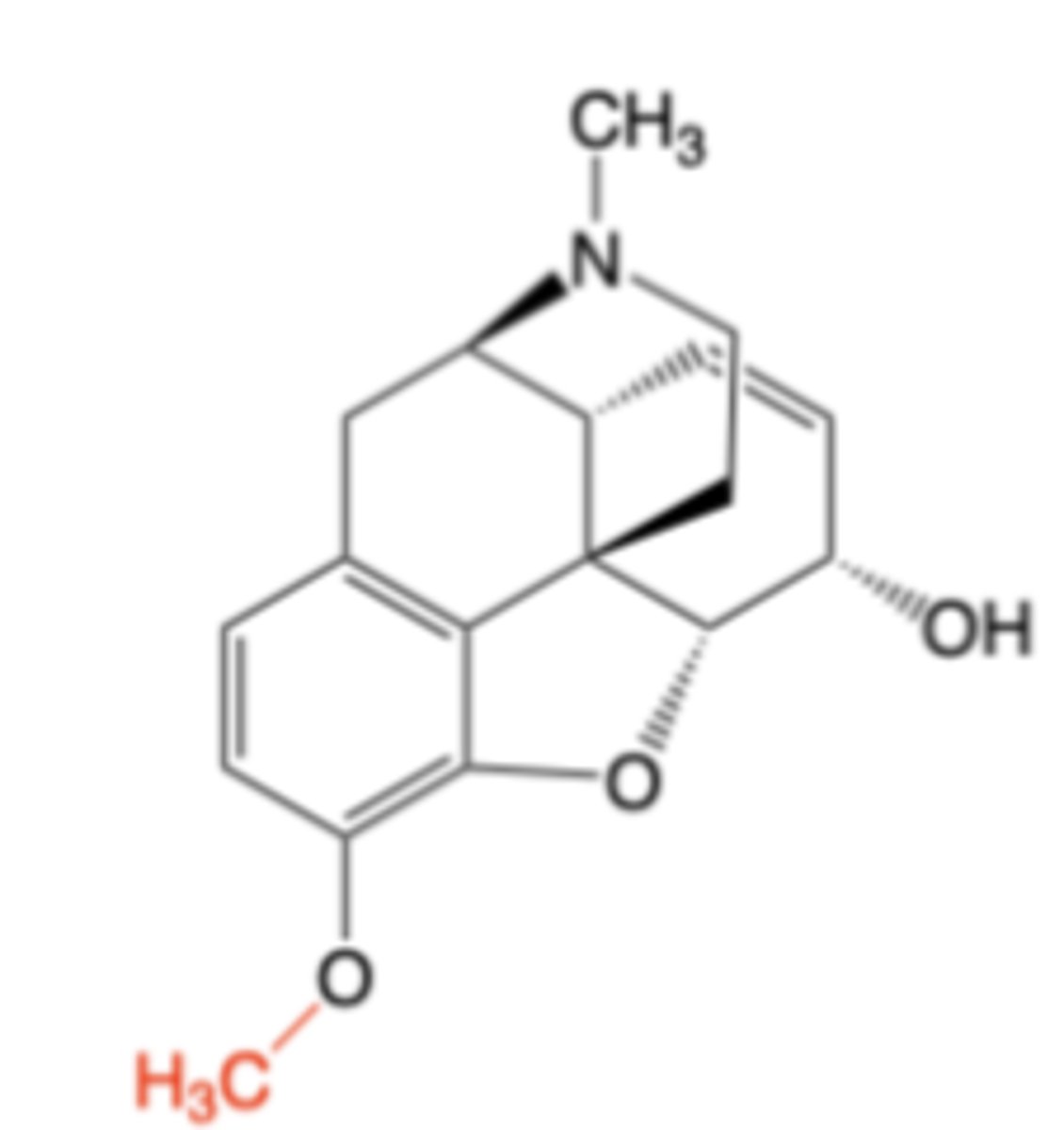
Hydrocodone
-Commonly co-formulated with acetaminophen, ibuprofen, or aspirin for oral analgesic
MOA: Selective mu-opioid receptor agonist -> similar in potency to morphine
-delayed onset of action due to delayed Tmax, high oral BA
ADME:
-Main metabolic pathways involve oxidation by either CYP3A4 or CYP2D6
-Absence of glucuronidation metabolism contributes to increase in oral bioavailability (Genetic polymorphism of CYP2D6 leads to less variable response and greater tolerability as compared to codeine)
Active metabolite: hydromorphone (CYP2D6 oxidation) --> analgesic activity
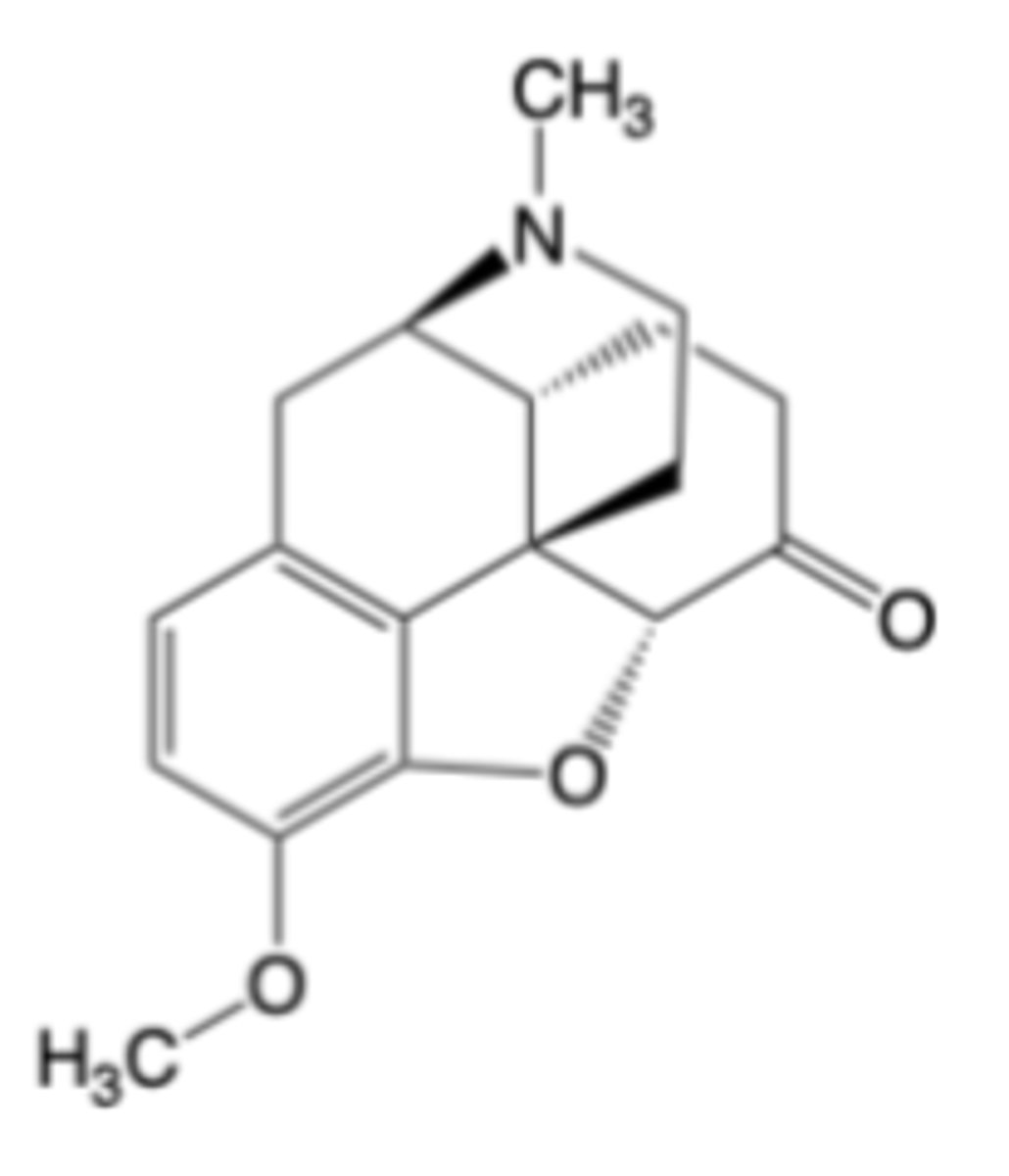
Oxycodone
-Single agent mainly available as an extended-release oral formulation of the hydrochloride salt (Oxycontin®)
-Co-formulated with acetaminophen (e.g., Percocet®) or aspirin (Percodan®) for use as an oral analgesic
MOA: Potent, selective mu-opioid receptor agonist -> ~5-10x more potent than morphine
mu Partial Agonists/kappa Modulators
• Buprenorphine
• Nalbuphine
• Butorphanol
• Pentazocine
mu Partial Agonists/kappa Modulators
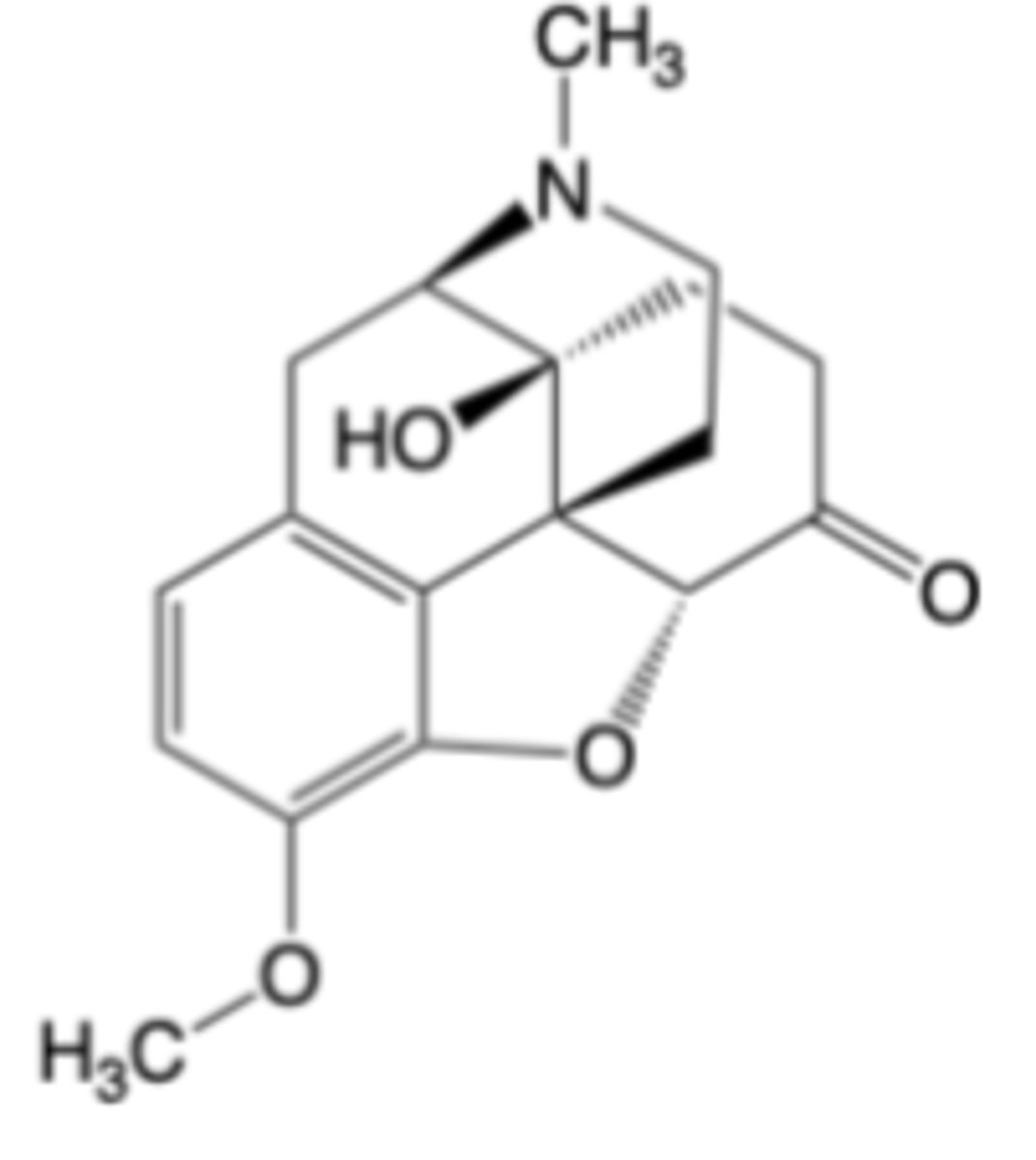
Nalbuphine
-Relief of moderate to severe pain, supplement to anesthesia, analgesia in postoperative and obstetric settings
-Ceiling effect on analgesia and respiratory depression viewed as advantages (vs. mu full agonists) that reduce overdose risk
-MOA: mu partial agonist, kappa full agonist
-AEs: Sedation, respiratory depression, nausea, constipation, and dysphoria
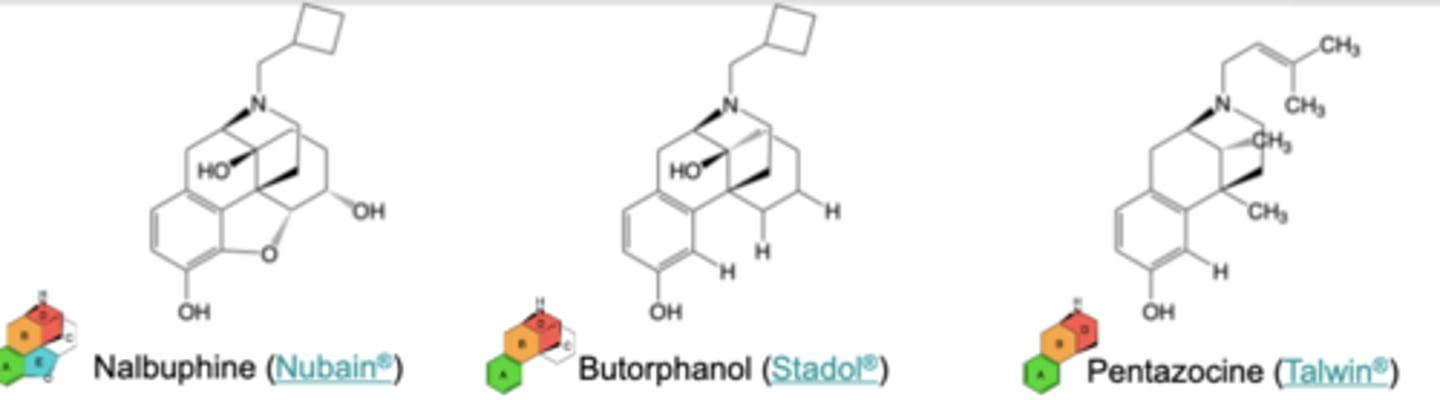
Buprenorphine
-Indicated for the treatment of moderate to severe opioid use disorder
MOA: Extremely potent, partial mu-opioid receptor agonist (~30-100x more analgesic potency compared to morphine) kappa-opioid receptor antagonist and opioid receptor-like 1 (ORL-1) agonist
-low (pseudo-irreversible) dissociation from mu-opioid receptors -> minimizes withdrawal symptoms
Oral administration of buprenorphine is generally avoided due to significant, first pass CYP3A4 oxidation (to norbuprenorphine) and glucuronidation (buccal, transdermal)
CI: acute or severe bronchial asthma, GI obstruction
Warnings: respiratory depression, QTc prolongation/severe hypotension, intracranial pressure/brain tumors/head injury/impaired consciousness
DDIs: Benzodiazepines (and other CNS depressants); CYP3A4 inducers or inhibitors can alter buprenorphine plasma levels
“Flexible” mu Agonists
• Fentanyl
• Sufentanil
• Alfentanyl
• Remifentanil
• Meperidine
• Methadone
• Tramadol
• Tapentadol
• Oliceridine
Oxymorphone and fentanyl bind to the endogenous agonist binding site of the mu opioid receptor
-both have important binding interactions with Asp147 and His297
-Oxymorphone -> Asn230 H-bond is potency enhancing
-Fentanyl -> Multiple additional pi stacking and lipophilic interactions contribute to enhanced potency (vs. morphine)
"Flexible" mu Agonists
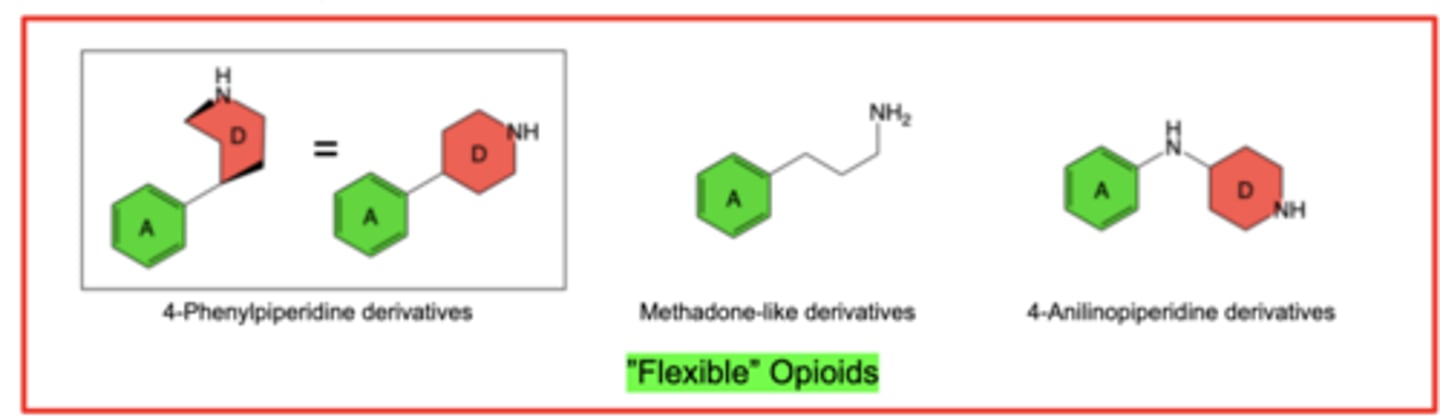
In general, efficacious 4-anilidopiperidine derivatives are more lipophilic than morphine congeners -> promotes central (brain) penetration
R1:
-Alkyl groups larger than methyl (-CH3) promote binding, with N-(2-aryl)ethyl being optimal for agonism in fentanyl analogs
R2:
-R2 = carboxymethyl (-CO2CH3) or methoxymethyl (-CH2OCH3) can sharply increase binding and agonism
R3:
-Small alkyl substituents can enhance μ- binding as well as selectivity over δ- and κ-receptors
Structure-Activity Relationships (SAR) of 4-Anilinopiperidine Agonists
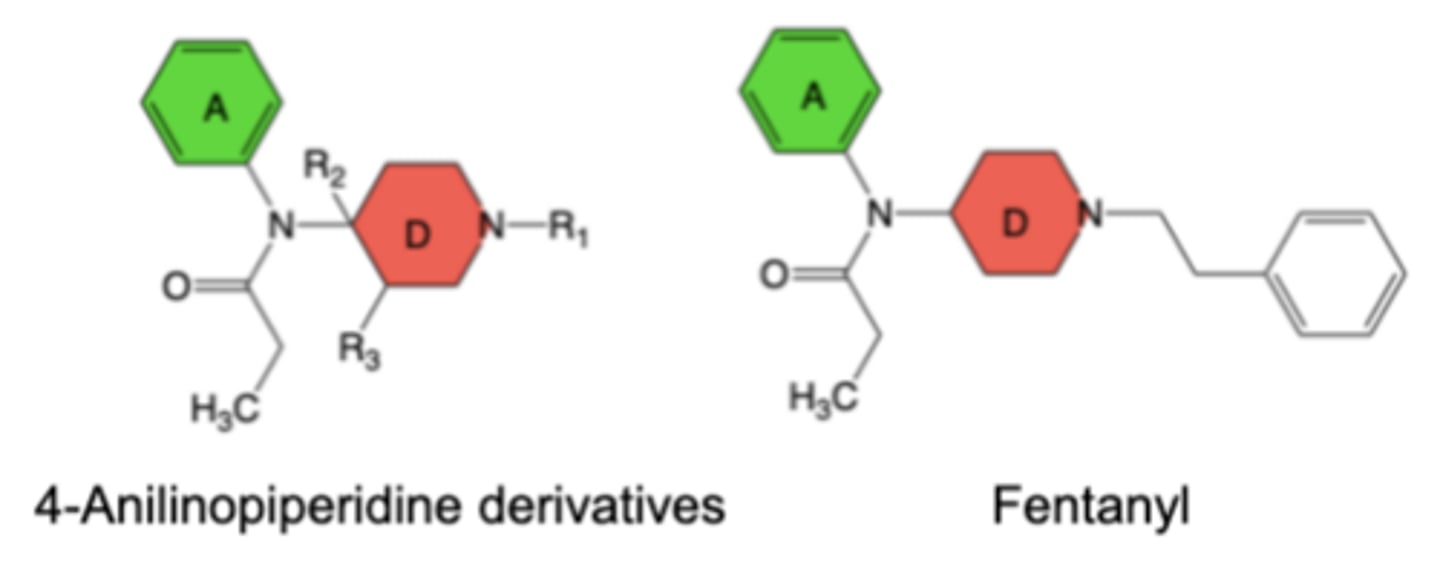
Fentanyl
-Available in formulations of its citric acid salt for injectable (for short duration during anesthetic periods, pre-medication, and maintenance in post op)and free base for transdermal extended-release (for management of severe chronic pain and cancer pain)
-BBW: risk of addiction, abuse, and misuse that can lead to overdose and death
MOA: Extremely potent, fast-acting selective mu-opioid receptor agonist -> ~100x more potent than morphine
Very short onset of action
CYP3A4 oxidation (N-dealkylation) to norfentanyl (inactive) is primary metabolic process
CI: hypersensitivity, impaired pulomary function
Life-threatening risks: addiciton, abuse, and misuse, respiratory depression, concomitant use with benzodiazepines or CNS depressants, serotonergic drugs, CYP3A4 inhibitors
AEs: Constipation, nausea, somnolence, lightheadedness, dizziness, sedation, vomiting, sweating
Fentanyl's greater lipophilicity promotes more rapid entrance into rat brain compared to morphine
Fentanyl is considered a “high-efficacy” agonist, while morphine is a “lower-efficacy” agonist
Fentanyl’s mu-receptor EC50 is ~10-100x lower (more potent) than morphine’s EC50
Fentanyl’s extensive mu-receptor binding interactions induce more pronounced conformational changes associated with full activation as compared to morphine ->linked to a higher intrinsic efficacy for fentanyl
Fentanyl maintains its ability to produce maximal agonist effects even when a large portion of mu-receptors are blocked or inactivated -> morphine can’t do this
Fentanyl vs. Morphine: Key Differences
Meperidine (Demerol)
-MOA: Selective mu-opioid receptor agonist -> 0.1x as potent than morphine
-Mechanism-based very similar to morphine (fewer GI issues -> lower incidence of constipation)
-More sensitive to CYP3A4-based drug-drug interactions than morphine
-Greater risk of neurotoxicity (seizures, myoclonus, tremors) due to normepiridine accumulation
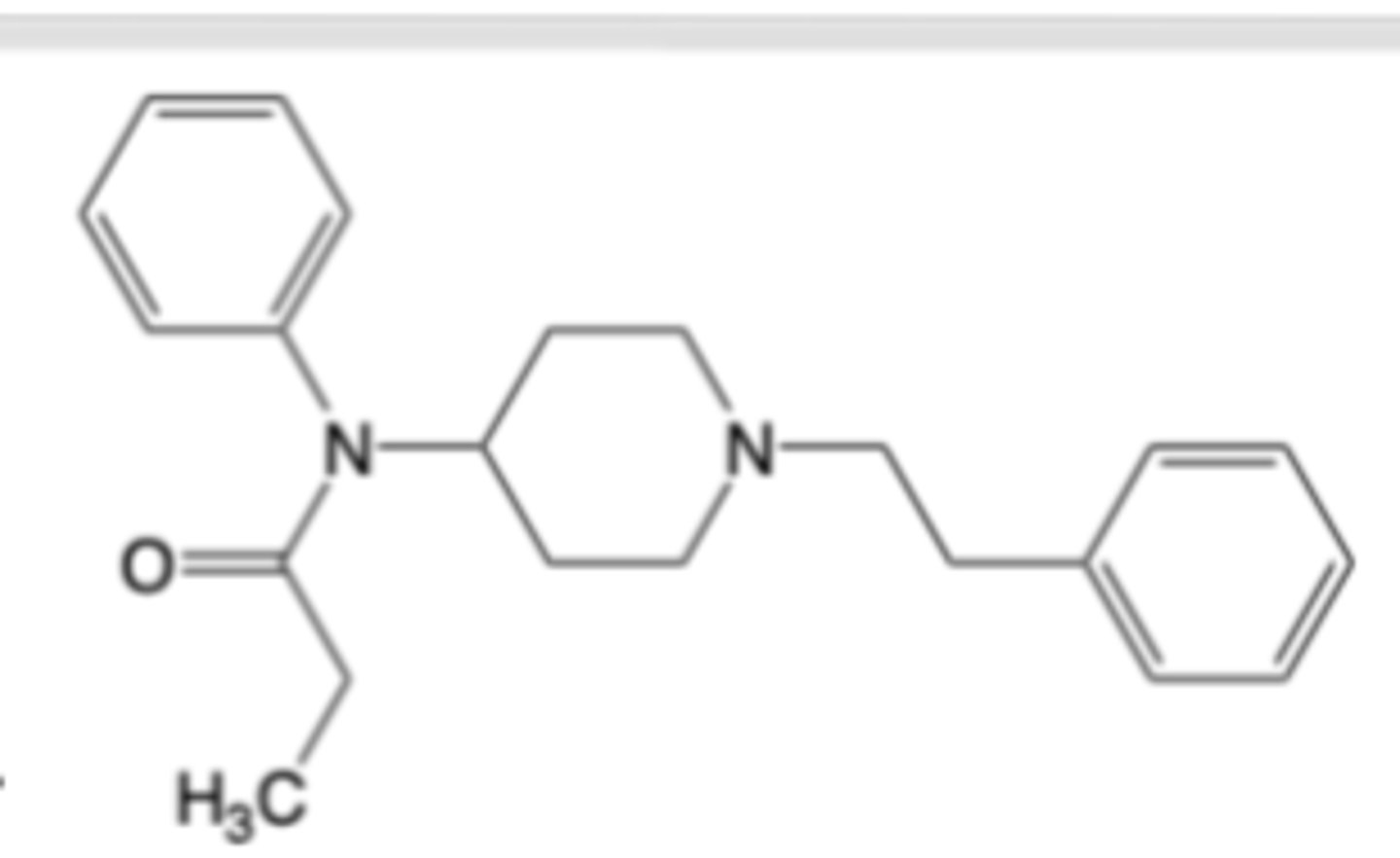
Methadone (Dolophine®)
-Extra indication: Indicated for detoxification treatment and/or maintenance treatment of opioid addiction (heroin or other morphine-like drugs)
-Oral
LIMITATIONS OF USE: Methadone products used for the treatment of opioid addiction in detoxification or maintenance programs are subject to the conditions for distribution and use required under 42 CFR 8.12
MOA:
-R-enantiomer is a selective mu-opioid receptor agonist -> similar in potency to morphine
-S-enantiomer is an N-methyl D-aspartate (NMDA) receptor antagonist -> added neuropathic analgesia via inhibition of serotonin and norepinephrine reuptake
Long t1/2
Same CI risks and AEs as fentanyl with more DDIs with CYPs
DDIs: Antiretroviral agents (CYP3A4 inhibitors), arrthymia-inducing drugs, mixed agonist/antagonist opioids, MAO inhibitors
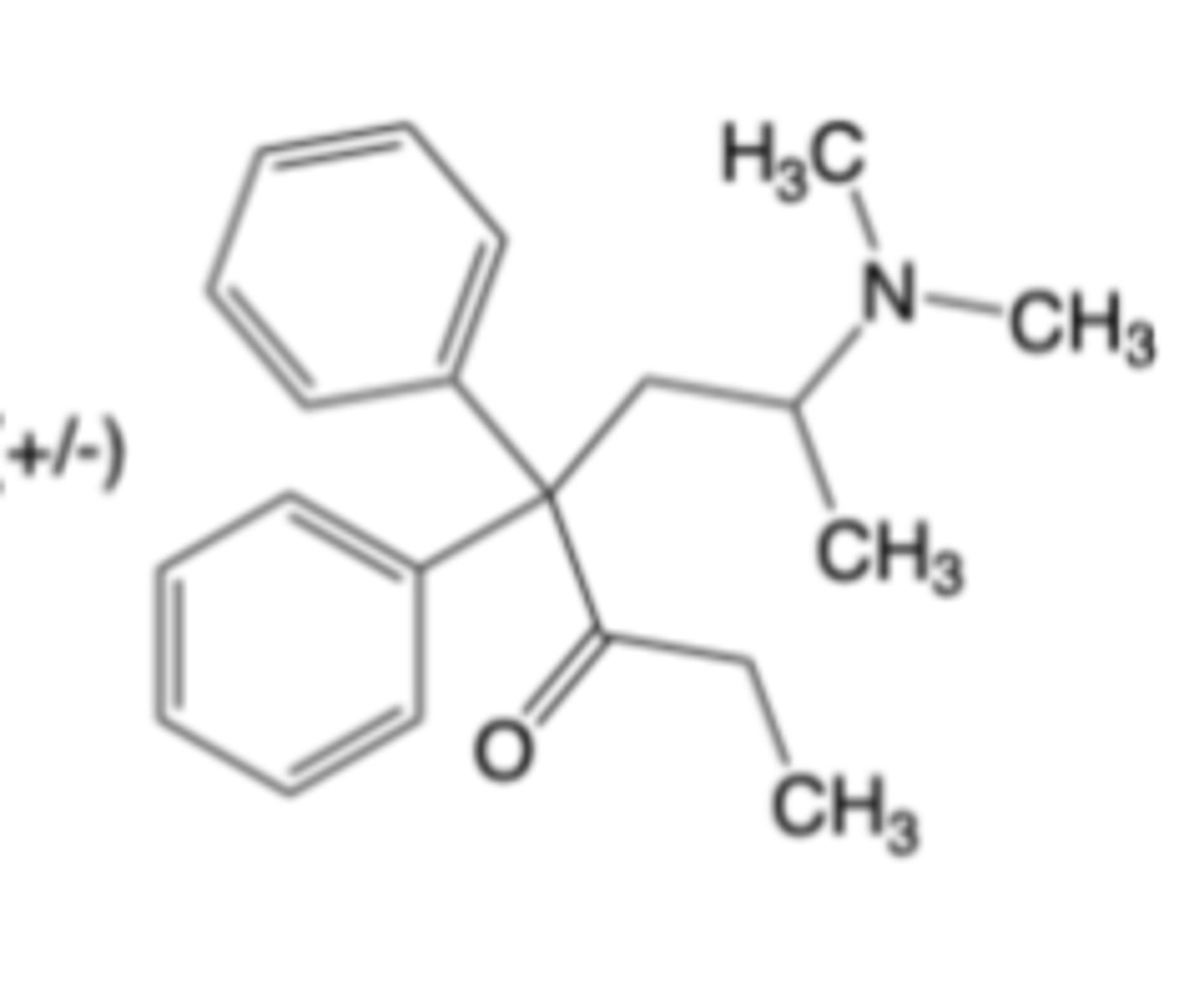
Tramadol
-MOA: mu-opioid receptor agonism and monoamine reuptake inhibition (NE reuptake inhibition, 5-HT reuptake inhibition
-enantiomers
-~0.2x as potent than morphine
-Warning for serotonin syndrome, risk of seizsure
-CYP DDIs
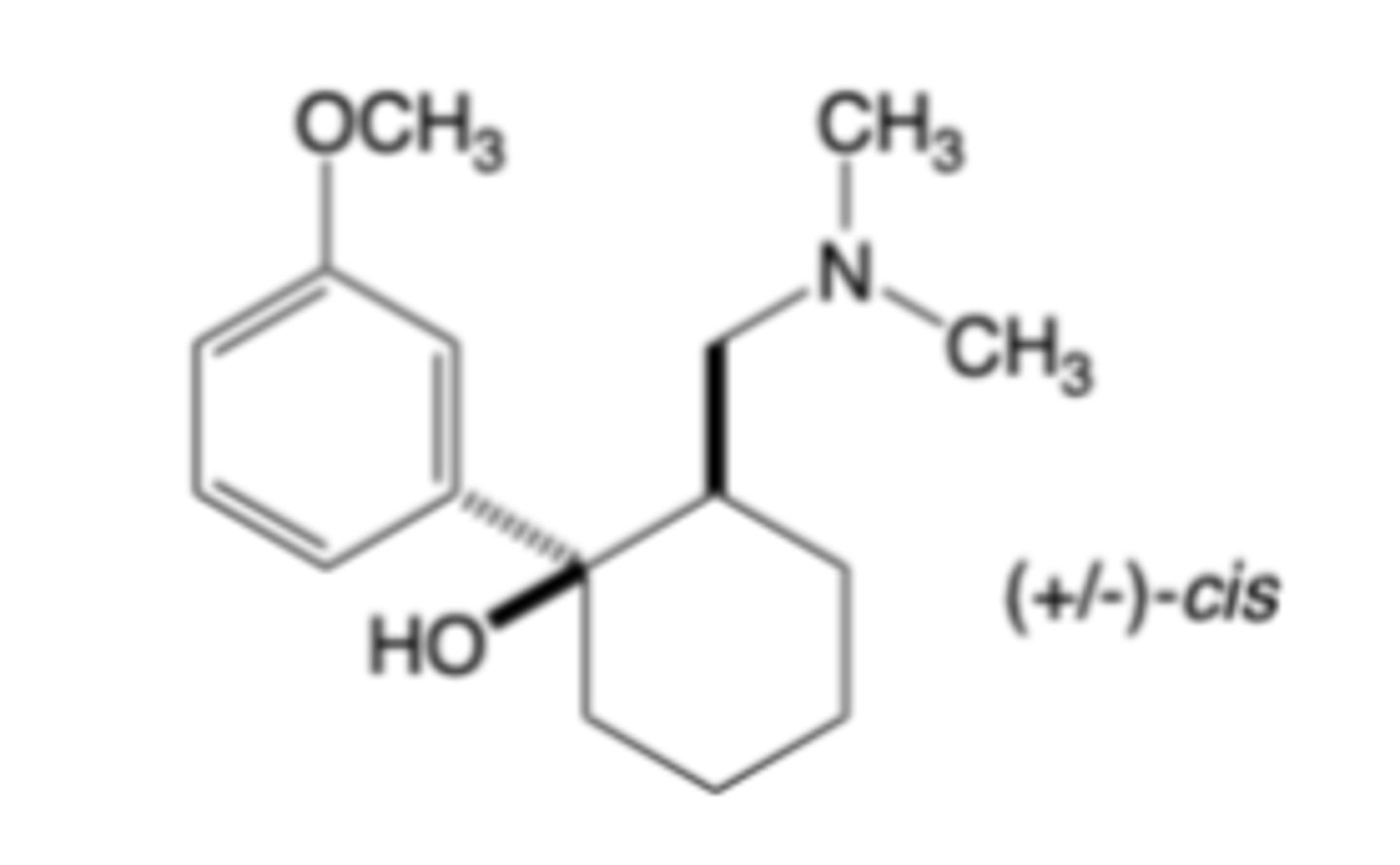
Tapentadol
-MOA: mu-Opioid receptor agonism and norepinephrine reuptake inhibition -> 0.4x-1x as potent than morphine
-Phase 2 metabolized (less DDI than tramadol)
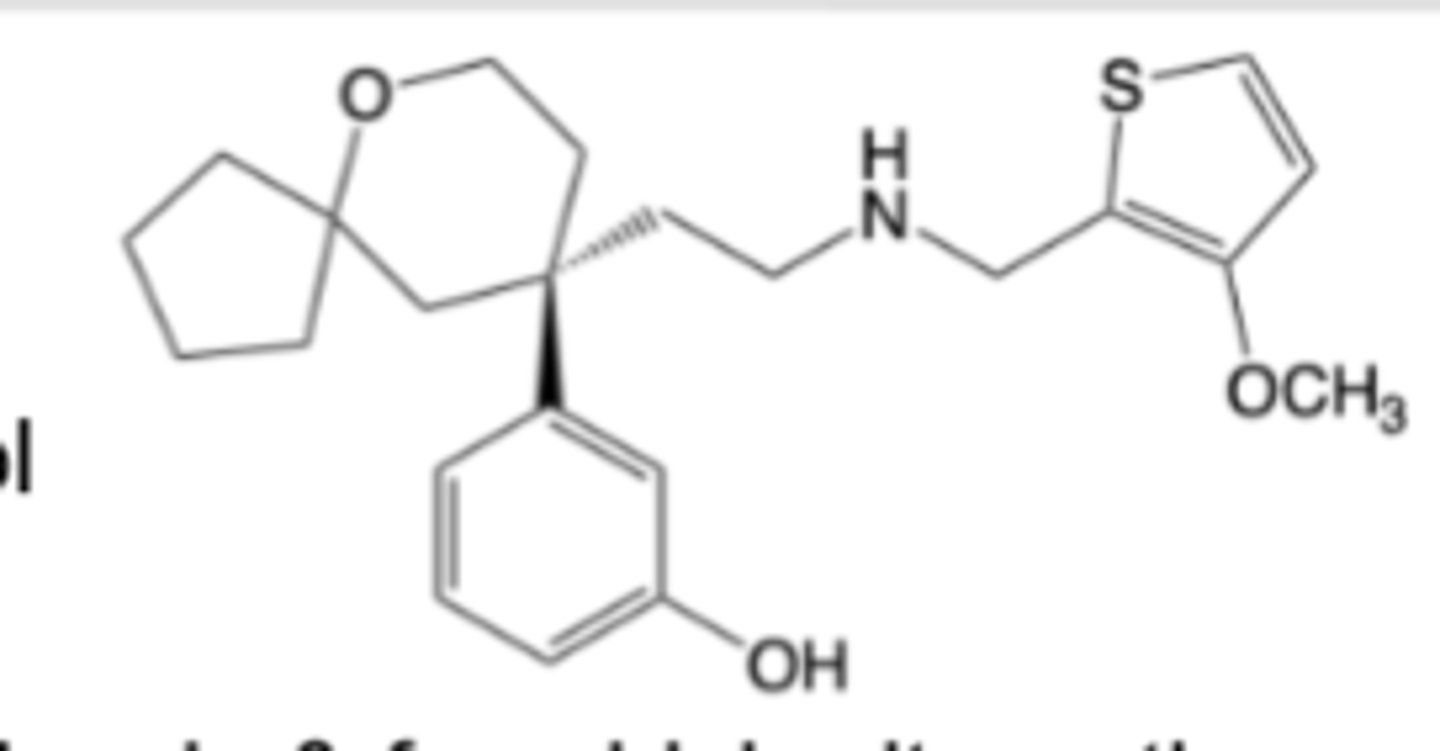
Oliceridine
-MOA: Biased mu-opioid receptor agonist (engages G-protein, but not beta-arrestin) -> analgesic potency similar to morphine
-Reduced AEs in some patients compared to morphine
Loperamide (Imodium®)
-available OTC
-Indicated control and symptomatic relief of acute nonspecific diarrhea in patients >2 years of age
-Indicated for chronic diarrhea in adults associated with inflammatory bowel disease
-MOA: Mu-Opioid receptor agonism -> ~1x as potent as morphine
-Excellent substrate of the P-glycoprotein (pgp) efflux transporter which precludes crossing the blood-brain barrier
Warning: Risk of torsades de pointes, cardiac arrest, and death with higher than recommended doses
CYP or pgp inhibitors can cause toxicity
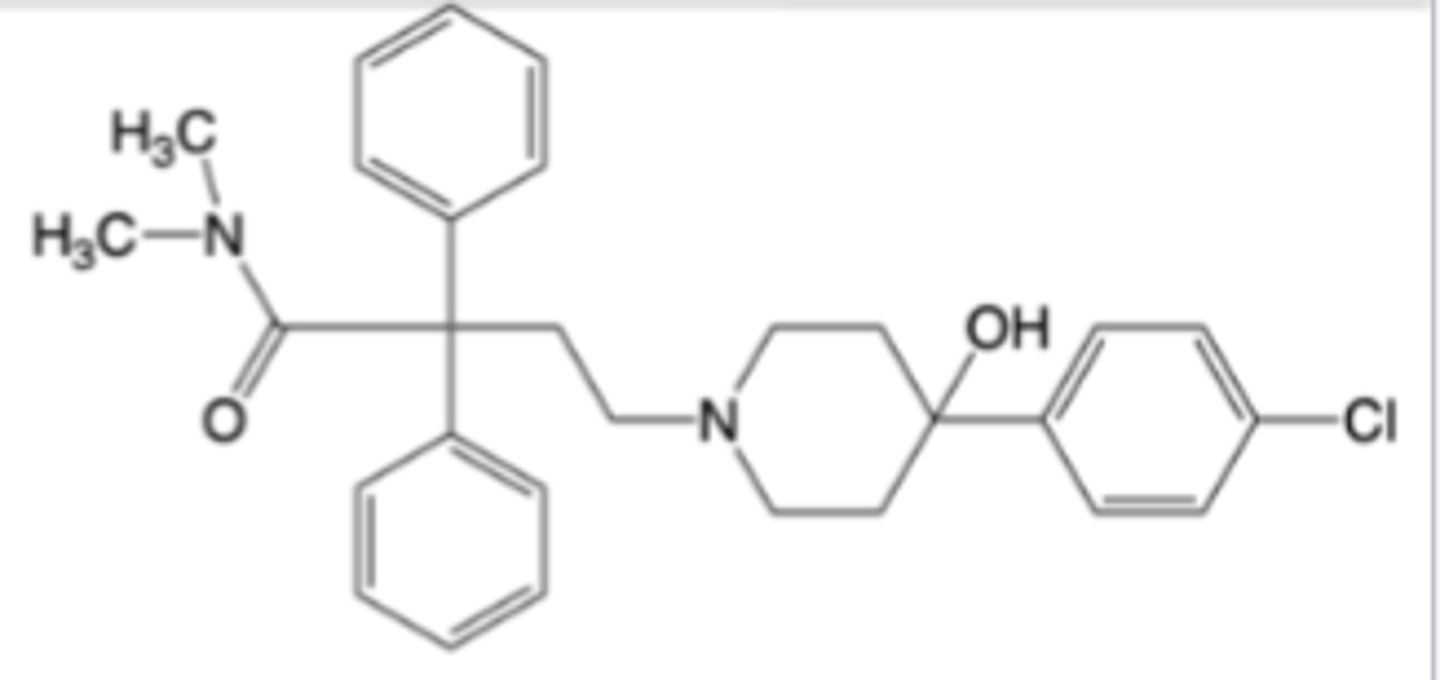
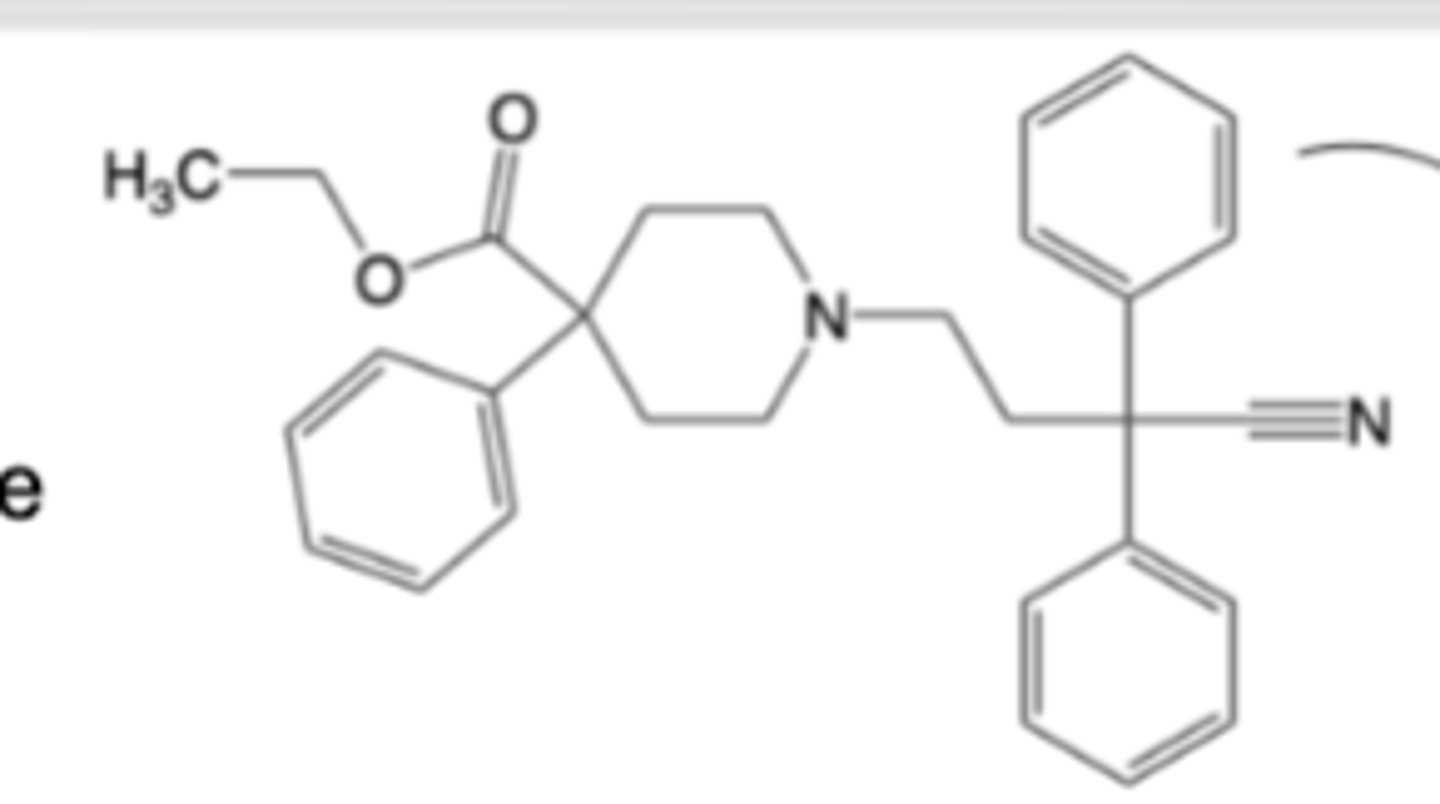
Diphenoxylate
-Co-formulated as the hydrochloride salt with atropine sulfate for oral administration
-adjunctive therapy in management of diarrhea
-Diphenoxylate is a prodrug of and rapidly converted to difenoxin on oral administration (Difenoxin is a weak mu-opioid receptor agonist -> much less active than morphine)
R1: Larger alkyl groups can bind well, but diminish agonism and/or introduce antagonist activity
Structure-Activity Relationships (SAR) of Mu Agonist Morphine Congeners
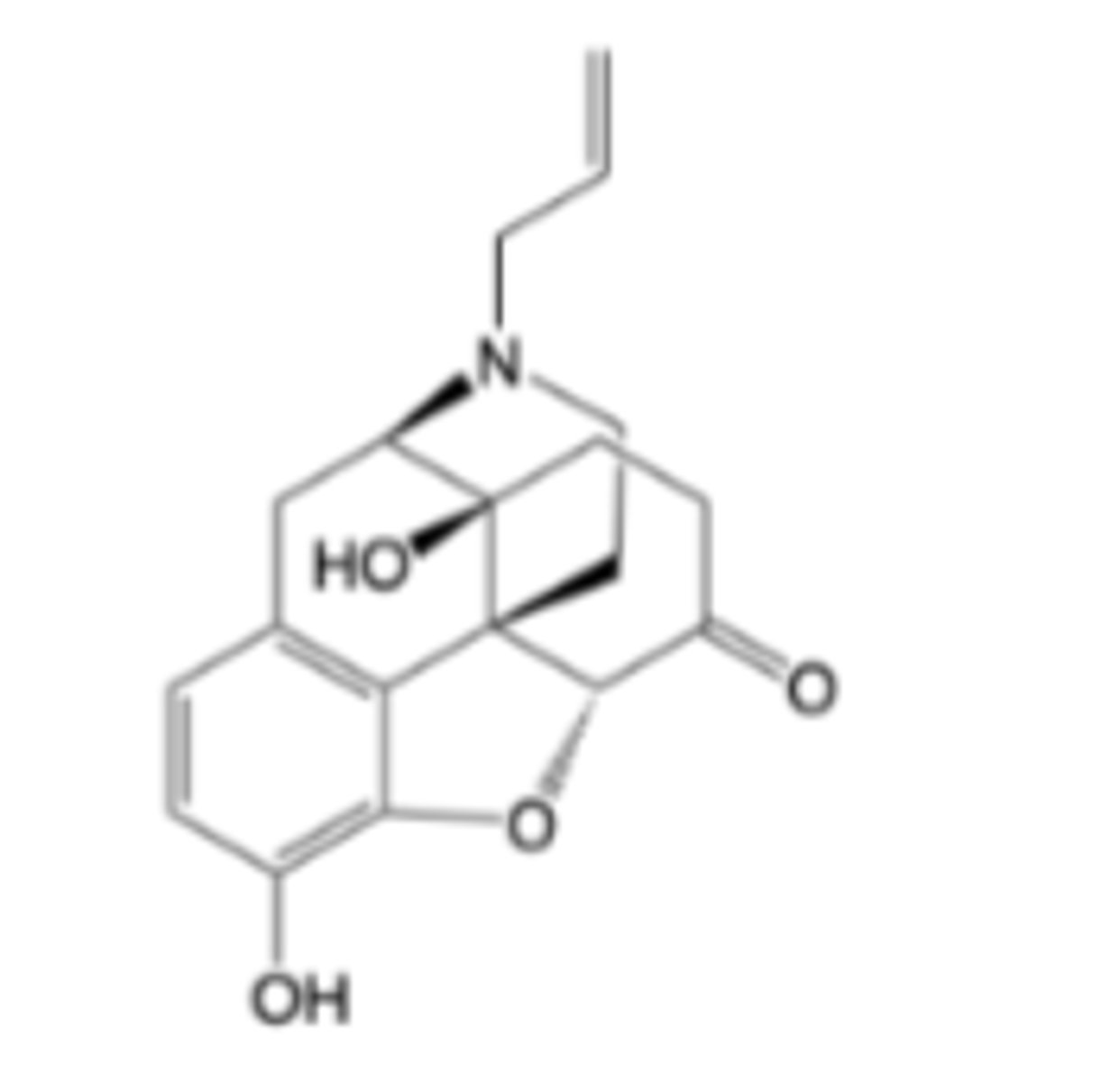
Naloxene
-Mu antagonist
-Nasal spray (Narcan) or subQ/IM injection syringe (Evzio)
-Emergency treatment of opioid overdose
-Very rapid onset, short duration of action
Risks: severe opioid withdrawal syndrome, ofcardiovascular complications in patients with heart disease, neonates, and postoperative patients
Metabolism primarily phase II
No DDIs --> single dose, short duration of action
Naltrexone
-Mu antagonist
-Treatment of alcohol dependence
-Prevention of relapse to opioid dependence
-Slow dissociation from mu receptor, long duration of action
Risk: severe opioid withdrawal syndrome, liver toxicity, injection site complications
DDIs: can counter effects of opioid-containing medicines (cough remedies, antidiarrheals, analgesics)
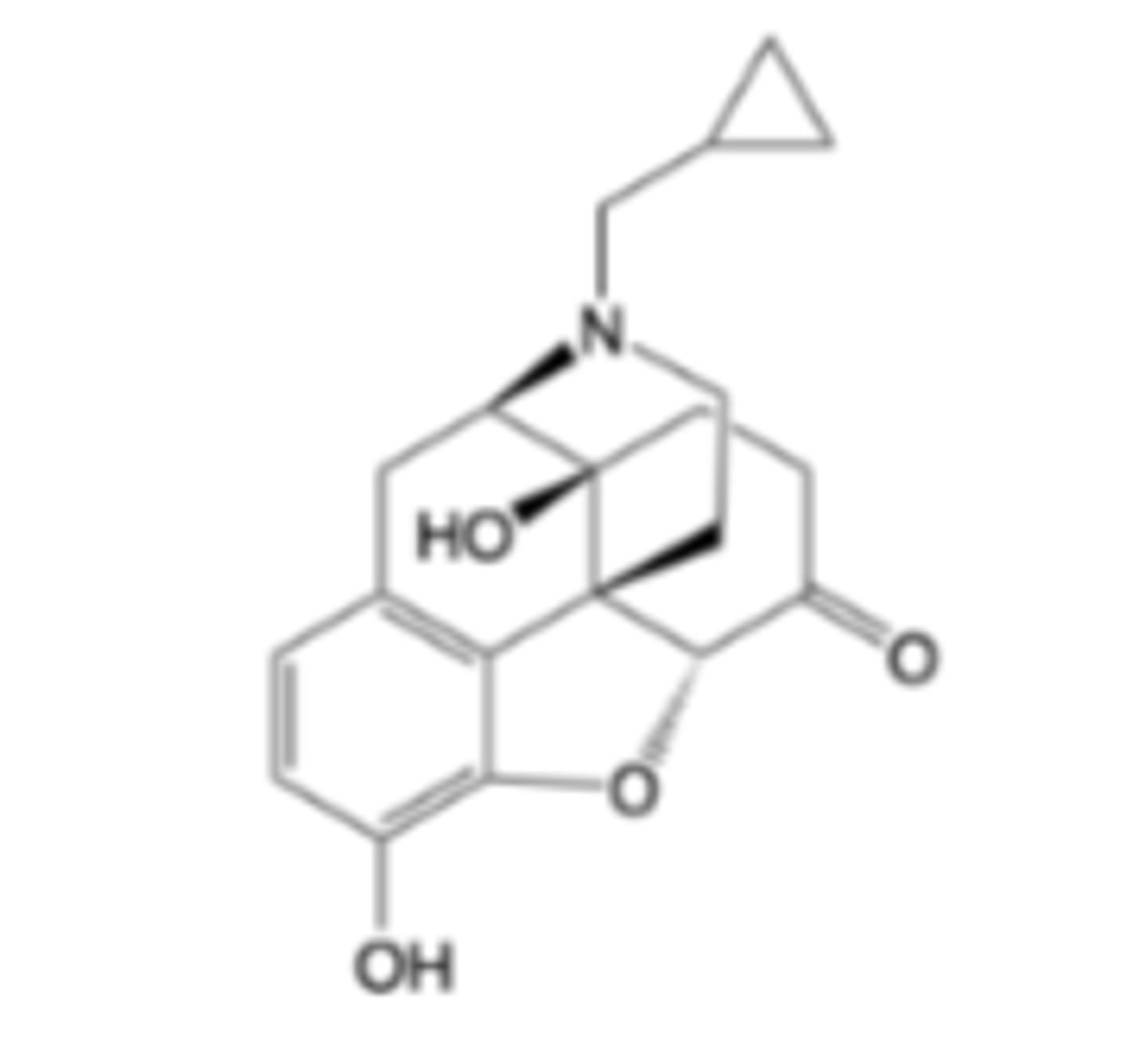
Methylnaltrexone bromide
-Mu antagonist
-Treatment of opioid-induced constipation
-Quaternary amine substructure prevents crossing blood-brain barrier -> does not interfere with analgesia
Warnings: Contraindicated in patients with known or suspected
mechanical gastrointestinal obstruction, Risk of opioid withdrawal syndrome, GI perforation
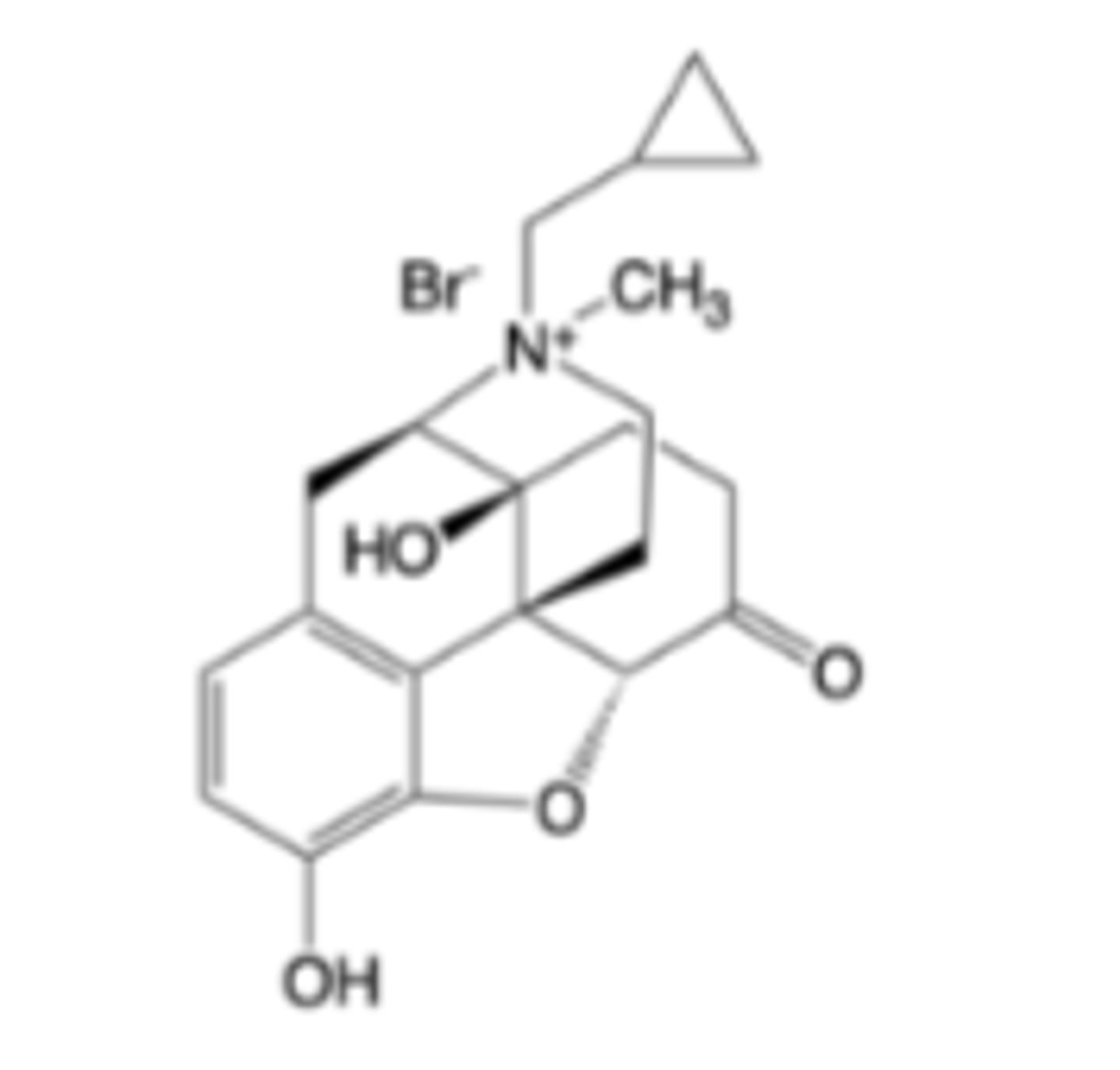
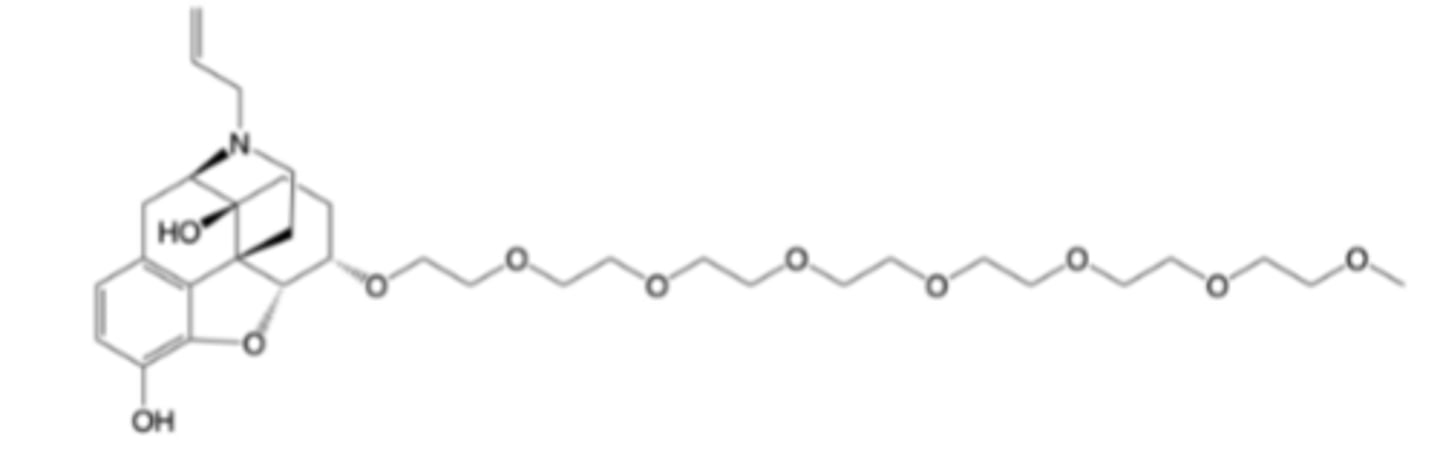
Naloxegol
-Mu antagonist
-Treatment of opioid-induced constipation
-Polyethylene glycol side chain and pgp sybstrate activity prevents crossing blood-brain barrier -> does not interfere with analgesia
Warnings: Contraindicated in patients with known or suspected
mechanical gastrointestinal obstruction, Risk of opioid withdrawal syndrome, GI perforation