Catalysis and Mechanisms
1/21
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
22 Terms
Define a catalyst mechanistically
A catalyst participates transiently in the reaction mechanism by forming and breaking bonds through one or more catalytic cycles, reducing ΔG(transition state) via enthalpic or entropic contributions, but is regenerated at end of each cycle.
A catalyst mechanistically should:
Stabilise the transition state relative to the reactant complex.
Control competing pathways (selectivity in the transition state being stabilised).
Outline the different representations of potential energy surfaces (3D, 2D, 1D and free energy) and what information each retains or loses.
3D potential energy surface:
Gives electronic energy of a system as a function of nuclear positions.
Easily locates stable intermediates, transition states (at saddle points).
Clearly shows topography for understanding dynamics.
Can explain why certain pathways are preferred.
2D potential energy surface:
Can give information on competing pathways.
1D potential energy surface:
How electronic energy changes along a single reaction coordinate.
Only gives lowest energy pathway.
Gives information of activation barriers, relative energies.
Free Energy Landscape:
Includes temperature dependent vibrational, rotational, translational contributions, which account for entropy too.
Describes how thermally accessible states are distributed.
Includes environmental effects including solvation and catalysis.
Explain why catalysis depends on free energy (ΔG ‡ ), not just electronic potential energy.
Electronic potential energy = enthalpic contributions.
Catalysts lower barriers not only by stabilising t.s. enthalpically but also by reducing entropic penalties e.g. via preorganisation.
How does concentration of reagents (1) and entropy (2) change the intrinsic barrier for a reaction.
changing concentration of the reagents does not change the intrinsic barrier. Increasing the collision frequency but does not change the energy required to reach the t.s.
will be impacted by reorganisational entropy. A highly ordered t.s. → high intrinsic barrier.
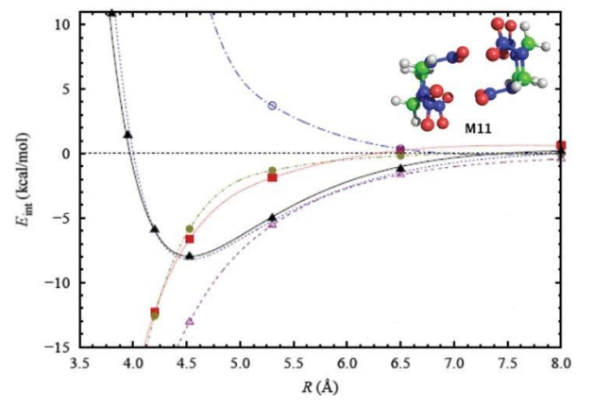
In this plot, identify the different SAPT energy terms.
Electrostatic: long-range interaction (observed at large R) and attractive (negative).
Exchange: repulsive, steeply rises at short distances. Rapidly → 0 at large R.
Induction: similar to electrostatics with more distance dependence.
Dispersion: always attractive, smooth decay, much shorter range.
Interaction = total of all energies. Exchange dominates at short range (repulsive) and dispersion/electrostatics at long-range (attractive)
What can activation strain analysis be used for in catalysis?
Computationally decomposes a reaction barrier between reactant and transition state in terms of the distortion of the reagents and the interaction energy between them.
Changes in the activation strain diagram with different catalysts can help explain whether a catalysts is lowering the energy barrier by lowering strain or by strengthening orbital interactions (or both).
Explain the principle of microscopic reversibility.
Says that the forward and reverse reactions share the same transition state. Landscape/pathway is always fixed for a fixed system.
Catalysts always affect both the forwards and backwards pathway.
Using Erying’s equation, how does changing ΔG ‡ affect rate quantitatively.
k = K (kBT/h)e-ΔG ‡/RT . The rate constant depends exponentially on the barrier height. Catalysts reduces the activation barrier (ΔG ‡) by decreasing the enthalpy of activation (stabilise t.s.) or increasing the entropy (reducing order).
Explain the idea of course-graining free energy barriers
Simplifying a complex multi-step free energy landscape into a reduced diagram. Removes many short-lived intermediates that don’t impact the macroscopic kinetics.
Replaces multiple T.S.s with one effective barrier.
How does Hammond’s postulate predict whether a TS resembles reactants or products + how can product influence rates through t.s. structure
Early t.s. -> looks like reactants.
Late t.s. -> looks like products.
The catalyst can tug on the kinetic barrier, from the product side, since the kinetic barrier has some resemblance to the product state. The more stable the product, the lower the preceding kinetic barrier, since the catalyst is already interacting before the TS
Give the Bell-Evans-Polanyi equation, defining each part.
ΔH‡ = aΔH0 + E0
Kinetic barrier = coefficient * reaction enthalpy * reaction energy/intrinsic barrier
Alpha = 1 → product like T.S.
Alpha = 0 → reactant like T.S.
(Note: can also be written in terms of G).
How can you distinguish transition-state stabilisation from ground-state destabilisation on a free-energy diagram.
2 different ways of reducing energy barrier ΔG‡ either by lowering the energy of the T.S. or raising the energy of the reactants.
Using SN1 and SN2 processes, explain how solvent properties can change ΔG‡.
SN2: polar aprotic solvent destabilises the reactant complex lowering the barrier.
SN1: polar protic solvent stabilises the intermediate, hence drags down the t.s. by Hammond's postulate. Lowers barrier.
Outline the difference between Pauli interactions (kinetic energy) and electrostatic interactions (Potential energy)
Pauli repulsions: energetic penalty for forcing two same-spin electrons into spatially similar regions. Electron density distorts to reduced overlap of filled orbitals, raising kinetic energy.
Coulomb attractions: electron density distorts to optimise electrostatic attractions i.e. minimising charge density.
Outline how Lewis-acid catalysed Diels Alder reaction is an example of Pauli repulsion in catalysis.
The Lewis acid binds to the carbonyl diene and pulls electrons away from the reacting centre, reducing orbital clashes and hence reducing kinetic penalties associated with orbital reconfiguration on collision
Increasing the Lewis acidity → activation barrier lowered.
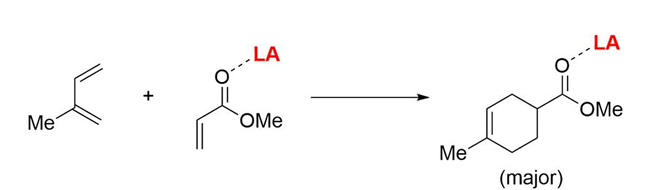
Outline the different contributions to the T.S. barrier
Orbital distortion: nuclei in molecules move to allow orbitals to adjust to T.S. Unfavourable.
Charge build-up: increased charge separation in T.S.
Pauli repulsions: ‘steric clash’
Orbital mixing: can stabilise the t.s. by charge delocalisation, pi backbonding, etc. or destabilise.
Statistical effects: probability of successful collisions.
Solvent effects
Draw normalised free-energy barrier diagrams that show reactions with better nucleophiles and more stable leaving groups. What is a good nucleophile and good LG?
Good nucleophiles = species with high energy HOMOs
Good LGs: species that from stable species (low-energy orbitals) when they depart.
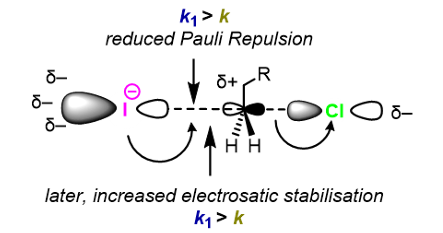
Explain how I- is a good kinetic nucleophile
The large, polarisable p orbital of I- (HOMO) distorts away from the C-X bond at long distances. Hence → reduced Pauli repulsion upon approach to the electrophile.
At short distances, electron density from the large, polarisable I- can easily flow into the virtual C-Cl sigma* orbital, increasing electrostatic stabilisation of the TS.
Outline ways in which the t.s. can be stablilised
Charge stabilisation via opposite charge, dipole or H-bond interactions.
Entropy/preorganisation - reducing total possible configurations, e.g. cyclic diene in Diels Alder → always in the s-cis conformation
Through-space polarisation & electric field effects e.g. reposition electrons to reduce steric/Pauli clashes.
Frontier MO tuning: reducing HOMO-LUMO gap e.g. LA to lower LUMO of the electrophile
Covalent activation: form a reversible covalent intermediate that changes in the reaction coordinate, lowering the effective barrier via a more favourable pathway e.g. using a nucleophilic catalyst