Alzheimer's Disease 1
1/28
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
29 Terms
Alzheimer’s
belongs to a larger category of diseases known as Dementia
46 million people diagnosed as of 2015, prediction that by 2050 there will be 131million
Not the only form of dementia (covers 65%), others like Huntington’s and some come from cerebrovascular disease (20%)
Most obvious effects are memory loss, decline in thinking, difficulties in communication and behavioural and personality changes
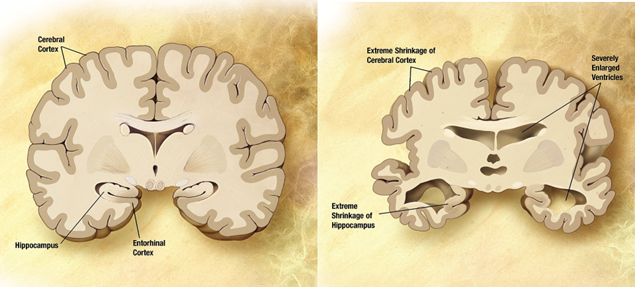
Prominent shrinking of the cerebral cortex, enlargement of the ventricles and disturbance of the hippocampus at the macro level
At the micro level shows abnormal neurons —> one of the hallmarks is conglomerates of protein in the EC space and accumulation of protein aggregates inside the cell
Protein aggregates
Outside the cell, large amyloid beta plaques
Plaques present in 100% of Alzheimer’s but can be found in the walls of the blood vessels in other diseases
Intracellular aggregates are tau tangles of (NFD) —> not exclusively inside the cell
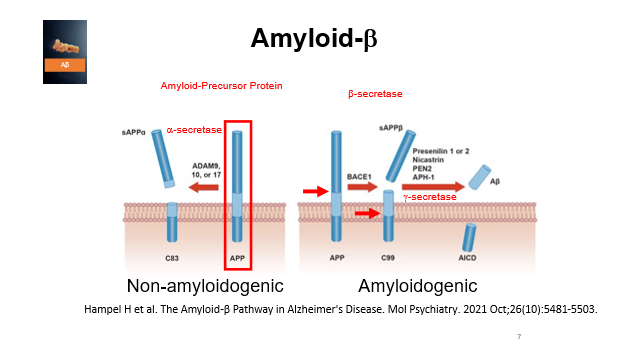
Amyloid beta
Key component is a plasma membrane protein APP (amyloid precursor protein) which is commonly expressed in neurons, in glial cells to a lesser extent
Normal brain function undergoes 2 types of processing
Non amyloidogenic
EC part of the protein cleaved right next to a membrane by a secretase (ADAM9,10 or 17) which is part of the alpha secretase family
This does not result in Alzheimers because the protein is soluble
Amyloidogenic
Beta secretase cuts of the EC part of the protein but in a different place, further out from the membrane
This allows the protein to be cleaved again, this time inside ethe membrane, by a gamma secretase complex
Results in AICD product which leads to the accumulation of AICD and A beta peptide
Usually these two processes both occur in normal neurons but in Alzheimers theres a shift in the balance of the amyloidogenic pathway leading to excess A beta
A beta
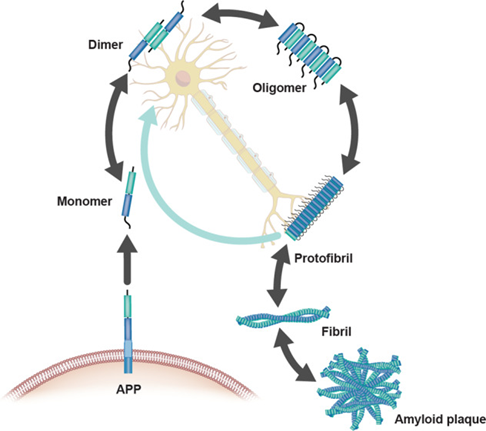
Initially released as a monomer but starts to clump as a dimer and oligomers forming aggregates
When aggregates the protein is highly insoluble and changes its conformation
What is the physiological role of APP in a non-disease state?
When APP is cleaved by an alpha secretase in the non-amyloidogenic pathway, it forms sAPPalpha (released outside the cell) and C83 (remains in the membrane)
sAPPalpha has several benefical roles in the brain including:
helping neurons resist stress and injury
enhances communication between neruons and supporting the learning process
promotes growth and repair of neuronal projections
stabilise neural activity
C83 fragment can be cleaved further by the gamma secretase, producing the p3 peptide which is not prone to aggregation and AICD which bnds to transcriptional complexes and actas as a co-regulator of transcription and act as an intracellular signalling molecule to influence
kinase pathways
calcium signalling
cytoskeletal organisation
Evidence linking Abeta with AD
cleavage of APP
Accumulation of APP
Cleavage of APP
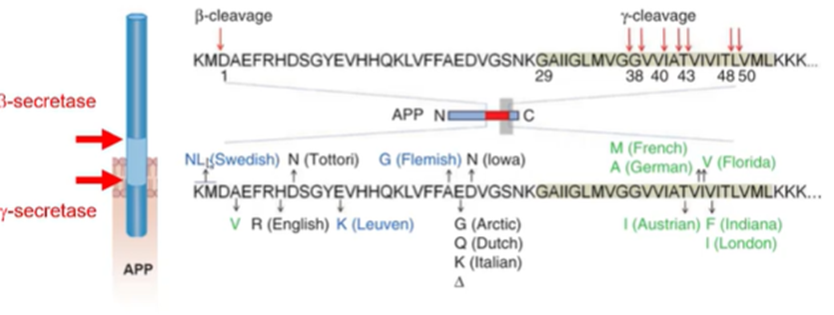
Genetic mutation in the APP (amyloid precursor protein) called the London mutation in the region that the gamma secretase cuts
Point mutation from V—>I
Definitively linked the production of Abeta with Alzheimer’s —> resulted in Alzheimer’s in 100% of cases
Further mutations identified but all mutations in the site that the gamma or beta secretase cleave the protein
Change in charge in the protein can make the secretase more or less efficient at protein cleavage
However, there have also been studies which show the opposite: mutations in the APP can also decrease the incidence of Alzheimer’s —> Icelandic mutation A673T resulted in an almost 50% reduction in amyloid beta production
Evidence linking Abeta with AD
cleavage of APP
Accumulation of APP
Accumulation of APP
APP is located on c21 which is additionally copied in downsynsdrome
Close to 100% of prevalence found in Down’s syndrome patients
Can release more A beta extracellularly due to additionally copy of APP which led to increased cleavage
Mouse models of AD
Race to create mouse models to understand the underlying mechanisms
Most mouse models based on overexpression of mutant proteins known to cause AD
Advantage of working with mice is that you can intervene with the pathology and generate transgenic models
We can also purify protein extracts from human patienst and insert it into the mice
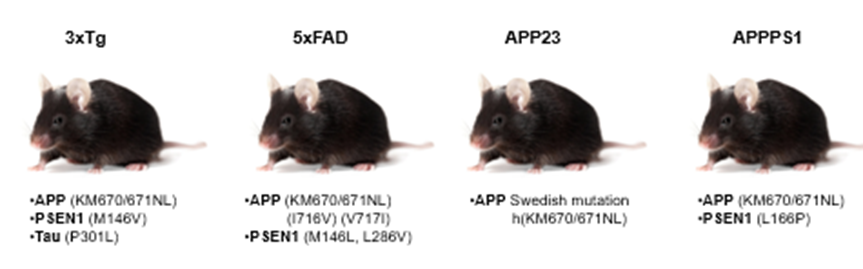
Types of mouse models:
APP23 one of the first mutants —> double AA change in APP protein (Swedish mutation
3xTg has mutations in APP, preseneline enzyme which forms part of the gamma secretase complex and Tau which leads to aggregation fof Tau
5xFAD is a very strong phenotype – has overexpression of APP with the Swedish mutation and London mutation, as well as 2 pathogenic mutation in presenilin
Human studies
can do post mortem studies, brain imaging, cognitive tests
Afetr this can do pathological analysis to identify amyloid and tau proteins
Pathology
amyloid plaques
synapse loss
compromised synapses
cognitive impairment
Cellular correlate of memory
Amyloid plaques
One of the hallmarks is the presence of Ab plaques
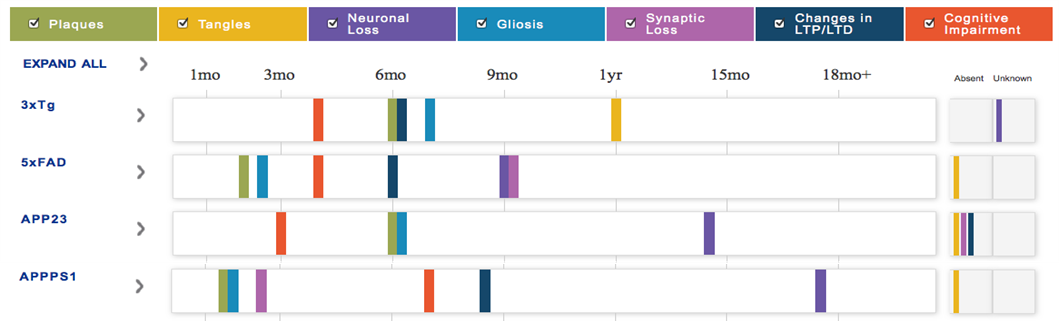
In both human and mouse studies, 3-month-old mice reflect humans in stage A of disease, 6 month old mice reflect humans in stage B and 9-month-old mice represent patients in stage C (for number of plaques)
Pathology
amyloid plaques
synapse loss
compromised synapses
cognitive impairment
Cellular correlate of memory
Synapse loss
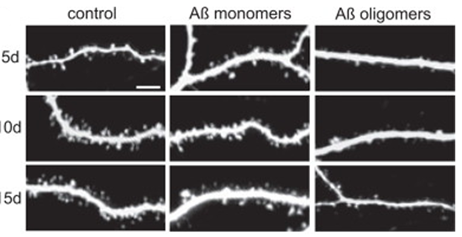
Slice of mouse brain hippocampus taken and incubated with dye to stain the neurons
In control cells the density of dendritic spines increases across 5-15 days
If we have the introduction of amyloid monomers (reflecting early stage Alzheimer’s), we do not change the number of dendritic spines
However, the oligomers have a huge affect on the dendritic spine density resulting in loss and fewer synapses
Pathology
amyloid plaques
synapse loss
compromised synapses
cognitive impairment
Cellular correlate of memory
Compromised synapses
In AD there are less synpases but they display dysfunction as well
Can take out the mouse hippocampus, the pathways in the hippocampus are very well characterised which allows you to stimulate these pathways with an alectrode and record the potential
APP Indiana mutation mice have less of a reduction in transmission in early stages (3-4wk and 2-4 month) but hugely increased in later stages
However, compared to controls, the fact that the transmission is decreased in these early stages, shows that there is something going wrong even before we are able to observe amyloid plaques and symptoms
Pathology
amyloid plaques
synapse loss
compromised synapses
cognitive impairment
Cellular correlate of memory
Cognitive impairment
Morris water maze: platform submerged in translucent water with visual cues around the room
Allow the mice to learn where the platform is and then a few days later, remove the platform and time how long they spend trying to find the platform
Mice with the Swedish/Indiana mutation shows much more random swimming patterns compared to the wildtype
Pathology
amyloid plaques
synapse loss
compromised synapses
cognitive impairment
Cellular correlate of memory
Long term potentiation – Cellular correlate of memory
Can be studies in the same way you study synaptic transmission: take out the CA3—>CA1 projections of the hippocampus and stimulate with an electrode and record with recording electrode
Stimulate at a high frequency to induce plasticity response
In human patients slices of brain taken and lysed then stain with an antibody for amyloid beta
All the Alzheimer’s patients had high Abeta and controls had little
Two types of Abeta: one that is high Mw (dimer), one that is low Mw (monomer)
Then leads us to think: can the cellular correlate of memory be affected differently by the monomers vs the dimers
The cellular correlate of memory (synaptic plasticity) is only decreased in patients with Alzheimer’s —> later proved that this was due to the dimers and not the monomers
Mouse models of AD
Even though 100% of patients show accumulation of A beta, in some mice models, you can start to see symptoms like cognitive impairment occurring before the incidence of plaque formation —> indicated that there is some signalling that something is wrong even before these plaques can be observed
All of them show cognitive impairments, but some of the other features like the formation of Tau tangles is not a common feature
Tau
Protein that usually exists associates with microbtubules in the axons
Usually controls the dynamics of the axon: stabilises and facilitates the growth of the axons in normal pateins
In AD tau is hyperphosphorylated which prevents it binding to the microtubules and starts to accumulate, forming neurofibular tangles
Shared pathology across other ND diseases like Parkinsons and temporal dementia
Evidence that links tau and Abeta in the pathogenesis
staining of mice brains
cerebral blood flow for brain activity
Morris water maze
Lifespan of mice
Staining of mice brains
Discovered a mutation in tau (P301L) which prevented the binding of tau to the microtubules which resulted in its aggregation and formation of tau tangles
Mutation induced the tau pathology which they visualised with an antibody stain: black signals in the soma can be observed which represents the accumulation of tau
In the Swedish mutant mice alone you don’t have this accumulation of tau
They then crossed this mouse with a transgenic mice with Swedish mutation (causing abeta aggregation) which resulted in this massive increase in tau tangles
Evidence that A beta potentiates the formation of Tau tangles
Evidence that links tau and Abeta in the pathogenesis
staining of mice brains
cerebral blood flow for brain activity
Morris water maze
Lifespan of mice
Cerebral blood flow for brain activity
Patients who did not yet have Alzheimers diagnosis but had pathological markers which resulted in mild cognitive impairments
The cerebral blood flow (proxy for activity in the brain, therefore neuronal function) measured via imaging —> only places where they observed abnormalities in blood flow/activity was the entorhinal cortex and parahippocampus
In mice models very early on before pathology expressed have the formation of these tau tangles but only in the mice which had the mutations in APP and the P301L mutation in the tau protein
Mice who only had the P301L tau mutation showed no early tau tangle expression
Evidence that links tau and Abeta in the pathogenesis
staining of mice brains
cerebral blood flow for brain activity
Morris water maze
Lifespan of mice
Morris water maze
Morris water maze experiments done in mice with wildtype tau and mice without tau showed that you can reverse the effects of APP mutations by removing Tau proteins
Tau mutations worsens the pathological phenotype but this can be reversed by the removal of tau
Not only can you see this with the Morris water maze memory test but also observe this with the cellular correlate of memory
In normal conditions, observe a potentiation of signal after repeated stimulation
With the amyloid beta protein this potentiation is depleted
If you use the amyloid protein but the reverse sequence, then interestingly, the potentiation effect resembles the control (normal plasticity) because it does not aggregate
after the removal of Tau you rescue the wildtype potentiation effect and show no cognitive effects even with the amyloid beta present
can reverse the cognitive and cellular impairments that is experienced with amyloid beta
These prove that tau is required to induce the mutated APP induced cognitive deficits and deficits in cellular correlates of memory
Evidence that links tau and Abeta in the pathogenesis
staining of mice brains
cerebral blood flow for brain activity
Morris water maze
Lifespan of mice
Lifespan of mice
Mice who have the amyloid beta and the tau mutation have a short lifespand and mortality rate of around 85% by 6 months
Not only do you get correction in memory with the removal of Tau but also get increased survival when you remove Tau
This also holds true when you express the Swedish mutation (APP23) alone without Tau —> increased lifespan of mice
Molecular targets and clinical trials
Eliminating the plaques is the main method of treatment —> whether if this is effective we don’t know
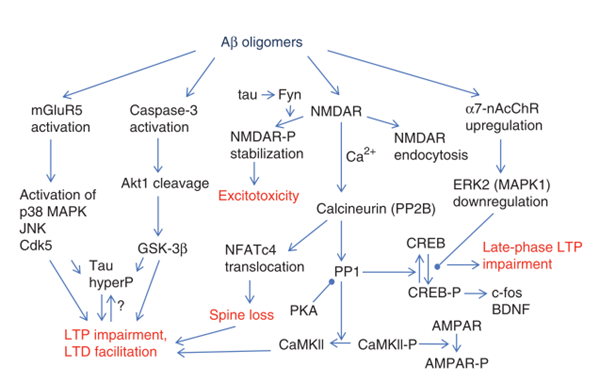
How does Abeta trigger the aggregation of tau?
Amyloid beta acts at NMDA receptors causing excess calcium influx
the excess calcium causes the activation of kinases that modify tau, particularly:
GSK-3beta
CDK5
MAPkinases
This causes the hyperphosphorylation of tau which leads to its aggregation and mislocalisation
Secondarily, the prescence of A beta plaques leads to the activation of microglia and astrocytes, who’s cytokines can also activate tau kinases
Furthermore, the ROS produces as a result of inflammation can drive the aggregation of tau
Vaccines
Injecting the amyloid proteins (Abeta1-42) and allowing antibodies to be generated in response
Plaques almost completely disappear from the mouse models and can rescue some of the cognitive deficits
Unsuccessful because the injection of the peptide, leading to the generation of antibodies —> leading to massive neuroinflammation resulting in death
Showed no improvement in cognition in human patients, inconsistent improvement in amyloid plaques —> some patients showed complete reduction, while others showed almost no improvement
This lead us to think that the removal of the amyloid plaques may not be able to rescue the cognitive impairment; however, this is caveated by the neuroinflammation which may prevent cognitive function in memory tests
Drugs
Biologics – introducing a molecule already existing in biology
Red = targeting amyloid, despite their previous failiures
Blue = targeting tau
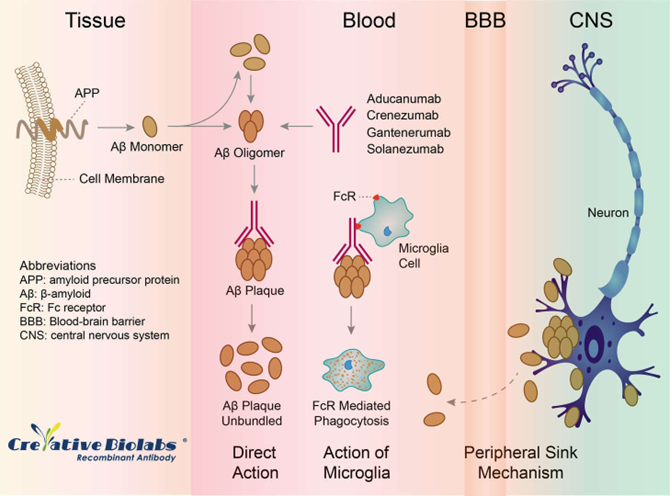
Solanezumab
Directly inject the antibody into the patient —> antibody is transient and present until it degrades
Binds only to monomers of Abeta —> even though monomers don’t cause the pathology, if you have a lot of monomers, eventually they will start to form oligomers —> acts as a sink of monomers
Compared to the vaccine is a passive immunotherapy
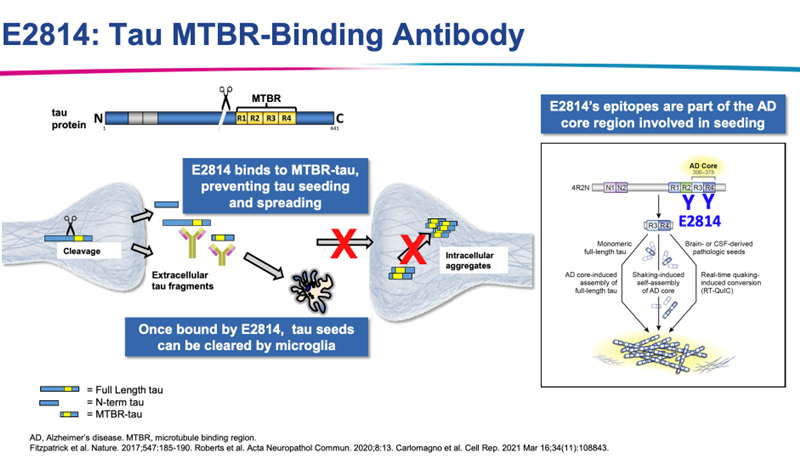
E2814
Directly inject the antibodies against Tau MTBR (microtubule binding region)
However, we know that mutated Tau is unable to bind to the microtubules, therefore, an antibody against the microtubule binding domain will only mimic the disease phenotype, so what is the rationale behind this?
Can only act in the blood or if it crosses the BBB can act in the EC space
Tau is also released into the EC space visa the nerve terminal and forms NFTs in the EC space
Can form toxic aggregates and also enter another cell and induce the pathology in another cell
Antibody binds to the region that usually the protein would use to bind to microtubules and sequesters Tau, prevents the formation of aggregates and also prevent the soluble Tau from entering the other cells
Passive immunotherapy, targets Ec fragments of Tau and prevents spread to other cells
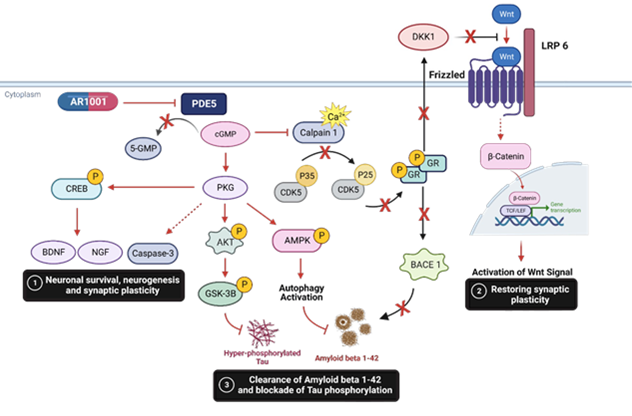
AR1001
Targets PDE5 —> is a phosphodiesterase 5 inhibitor
By inhibiting PDE5 you increase cGMP inside the cell
Increase cGMP in neurons leads to activation of pCREB, BDNF and NGF which promote neuronal survival, plasticity and neurogenesis
By boosting cGMP wou can also activate the autophagy pathway —> used to eliminate things that are abnormal inside the cell which clears the initial formation of neurofibular tangles or Abeta plaques
Also suspected to rescue memory loss phenotype and cognitive function
Summary of drugs
Almost all of these not yet approved
Nearly all of the drugs target the 2 hallmarks of Alzheimer’s: amyloid and Tau
Companies trying to develop antibodies which can not only recognise the monomers, but also the dimers, trimers and even plaques —> then seeing which one is most efficient
Also attempts to combine therapies which target both amyloid beta and tau
Used in very severe cases of AD where the formation of plaques is very high and Tau tangles high —> little chances of reversal
Currently, we don’t know if mild cognitive impairment is caused by the presence of plaques or there is another underlying cause for the impairment
In mild cognitive impairment, we know that these patients have a loss of synapses and synaptic function —> what if we try and target this aspect of the pathology and rescue this
Trends seen to target the earlier symptoms, attempts to see if they are more effective
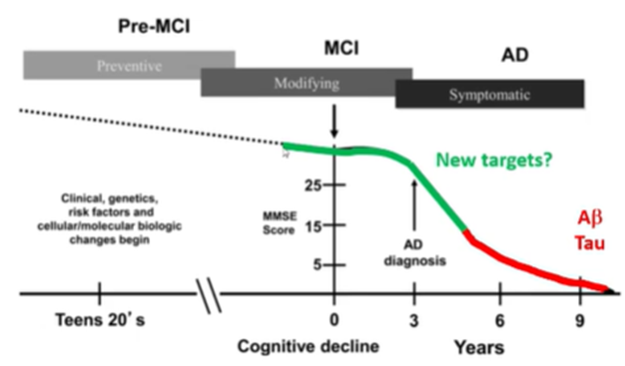
Targeting mild cognitive impairment
In order to do this we need to know what happens earlier on in these stages —> number of synapses decreases in the dentate gyrus and the CA1 region
Specific synaptic targets: we know that theres loss of plastcicity and synapse formation so we can target molecules known to be involved in this process
Example: Abeta oligomers activate NMDARs which causes the increase in Ca2+ into the cell which eventually leads to cell death —> can we target this?