PKPD Exam 3
1/44
Earn XP
Description and Tags
UBSOPPS
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
45 Terms
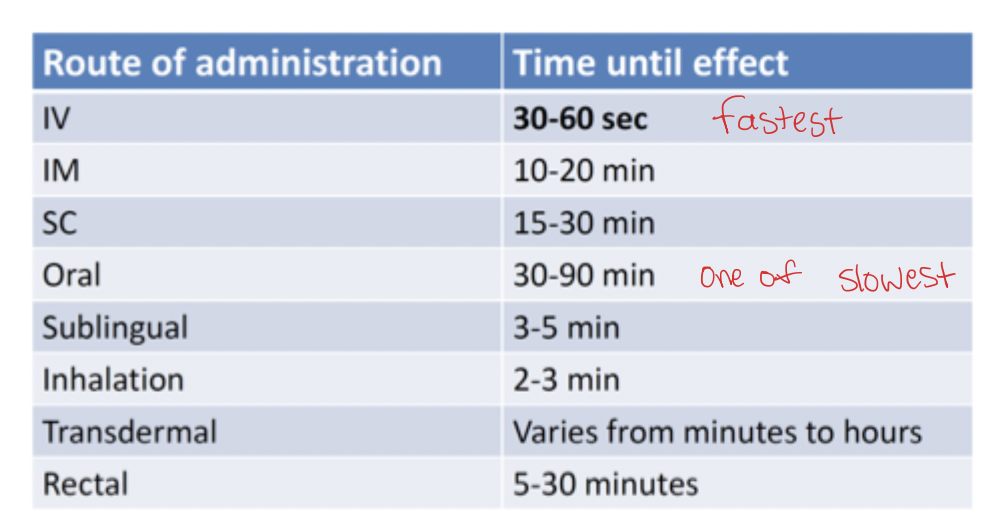
Time for Onset
IV → fastest
Inhalation
Sublingual
Rectal
IM & SC
Oral → one of the slowest
Transdermal
IV Route
Pros
all dose is delivered directly to blood stream
can do bolus or infusion in controlled amounts
Cons
Inconvenient and painful
Requires trained personnel
IM Route
Pros
Rapid Onset
Depot injections, larger than SC
Almost 100% bioavailability
Cons
Not large volumes
painful, inconvenient
site of injection can influence extent of absorption
SC Route
Pros
Slower constant absorption over longer period of time
Almost 100% bioavailability
Patient can inject themselves with minimal training.
Cons
Site of injection can influence extent of absorption
Can cause tissue damage
limited volumes
Intranasal Route
Pros
Ease of Administration
Rapid absorption and onset of action
Reduction of systemic side effects
Cons
Local irritation
lower bioavailability
efficiency depends on delivery system
Inhalation Route
Pros
Self administration
Large surface area for absorption
Reduction of systemic side effects.
Cons
Highly dependent on formulation
Highly dependent on delivery system
Sublingual & Buccal Routes
Pros
Avoids first pass metabolism
Rapid absorption and onset
Cons
Not suitable for large doses
Transdermal Routes
Pros
Ease of administration
Reduction of systemic side effects
Controlled prolonged delivery
Cons
Mainly only for local effects
Slow absorption
Low bioavailability
High dependence on drug characteristics such as logP, MW, and delivery system
Local irritation
Ocular Route
Pros
high drug concentration in localized area
self administration
Cons
limited types of drugs
not suitable for irritating drugs
Rectal Route
Pros
avoids first pass metabolism
useful for children, elderly, or unconscious patients
useful for vomiting or nausea
Cons
Invasive application
limited drugs can be administered this way
Oral Route
Pros
Convenient, cheap, painless
adjustable doses
self administered
Cons
Subject to first pass metabolism
Drug taste
PKPD of drug
Mechanism of Absorption
Drugs enter the apical side → Leave out the basolateral side
Transport
Passive Transcellular → high capacity, lipophilic, conc. gradient
Active Transcellular → substrate dependent, hydrophilic, active
Paracellular → low capacity, hydrophobic, conc. gradient
Transcytosis → active, very low capacity, macromolecules
Permeability vs Solubility Limited Absorption
If dissolution > absorption → most of the drug is dissolved before much is absorbed. Therefore permeability limits absorption
If absorption > dissolution → absorption cannot go any faster than the rate a drug is dissolved. Therefore absorption is dissolution rate limited
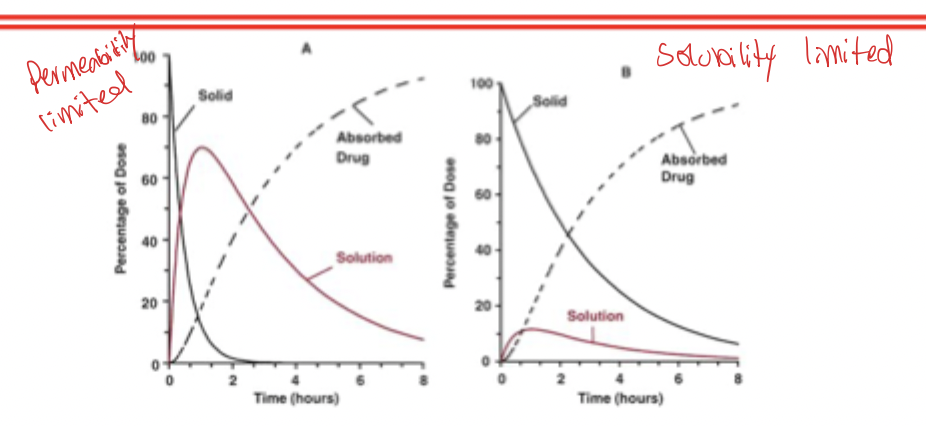
Kinetics of Absorption
Oral absorption of drugs is often first order kinetics
Absorption rate constant (ka)
corresponding absorption half life = (0.693 / ka)
Bioavailability
Fraction of dose that is absorbed into systemic circulation
No method using oral data alone
Absolute Bioavailability
comparing oral AUC data to IV AUC data
Relative Bioavailability
comparing two oral formulations
F can be greater than 1
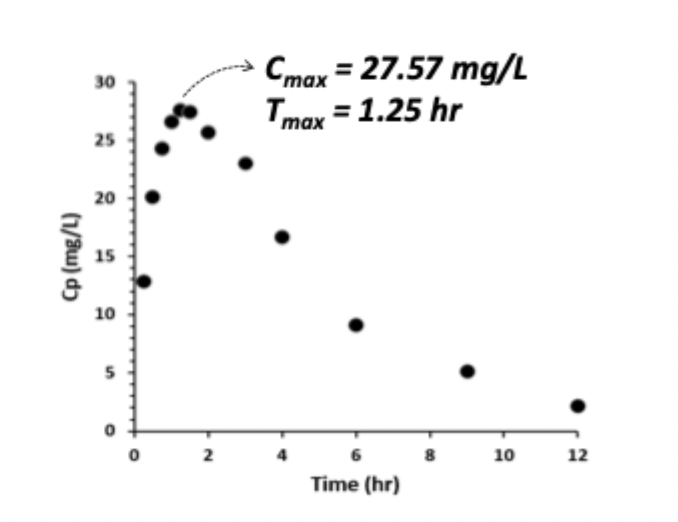
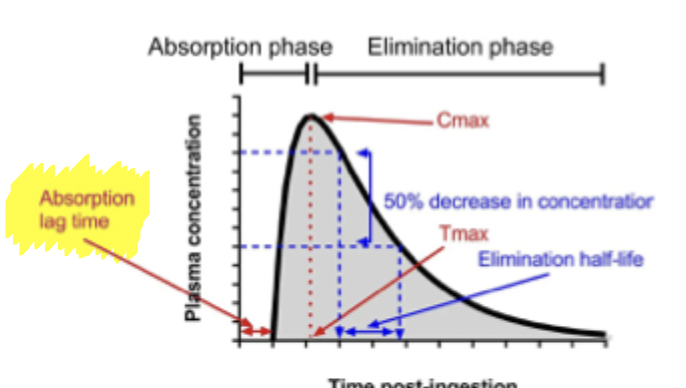
Peak Concentration
Cmax → peak plasma concentration
determined by D, F, Tmax (ka, Cl, V)
can calculate Cmax when ka >>> kel
Tmax → time to peak plasma concentration
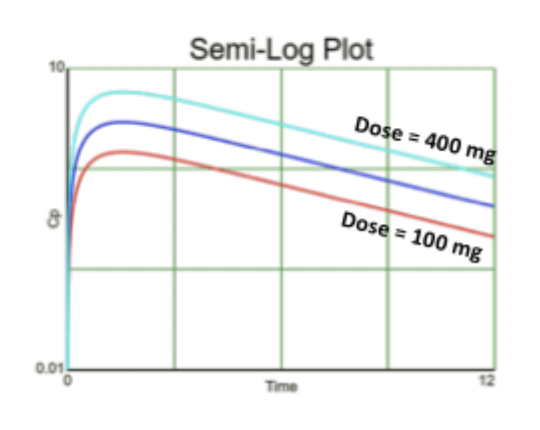
Effect of Dose on Graph
Tmax = ln(ka/kel) / ka - kel, AUC = F x dose / Cl, Cmax = D * F / V
If you increase dose:
Tmax stays the same
Cmax increases
AUC increases
Effect of F on Graph
Tmax = ln(ka/kel) / ka - kel, AUC = F x dose / Cl, Cmax = D * F / V
If you increase F (bioavailability)
AUC increases
Cmax increases
Tmax stays the same
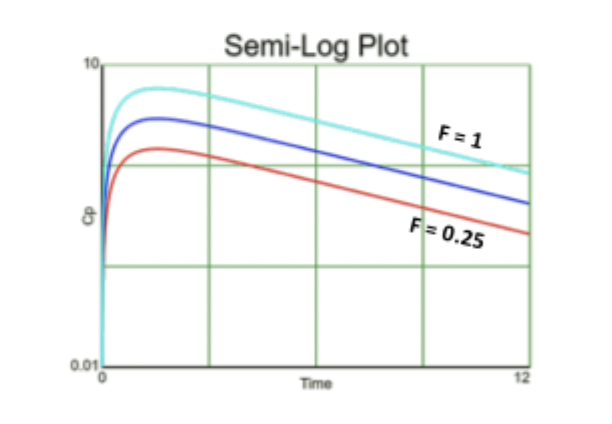
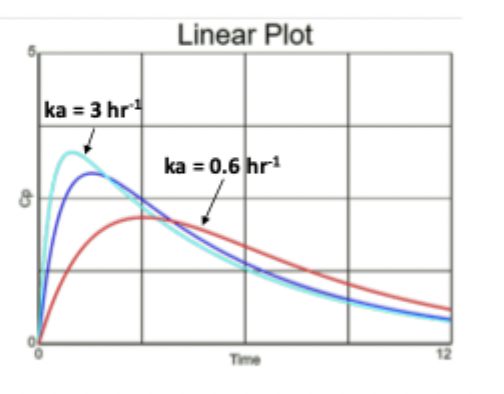
Effect of ka on Graph
Tmax = ln(ka/kel) / ka - kel, AUC = F x dose / Cl, Cmax = D * F / V
If ka increases (faster absorption)
AUC the same
Cmax increases, Tmax decreases
** think more drug absorbed all at once creating a spike on the graph, IV bolus-like **
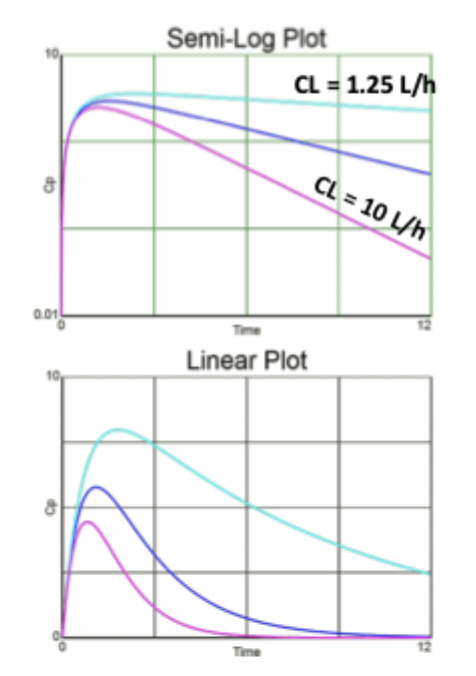
Effect of Cl on Graph
Tmax = ln(ka/kel) / ka - kel, AUC = F x dose / Cl, Cmax = D * F / V
If Cl increases:
AUC decreases
Cmax decreases
Tmax decreases
terminal t1/2 decreases
Flip Flop Kinetics
When ka < kel
drug absorption controls Cp and not elimination
sustained release products
terminal t1/2 increased
one dose has much larger amount of drug for entire day
should not be food sensitive
allows once daily formulations
AUC 0 → infinity = AUC 0 → tau at SS
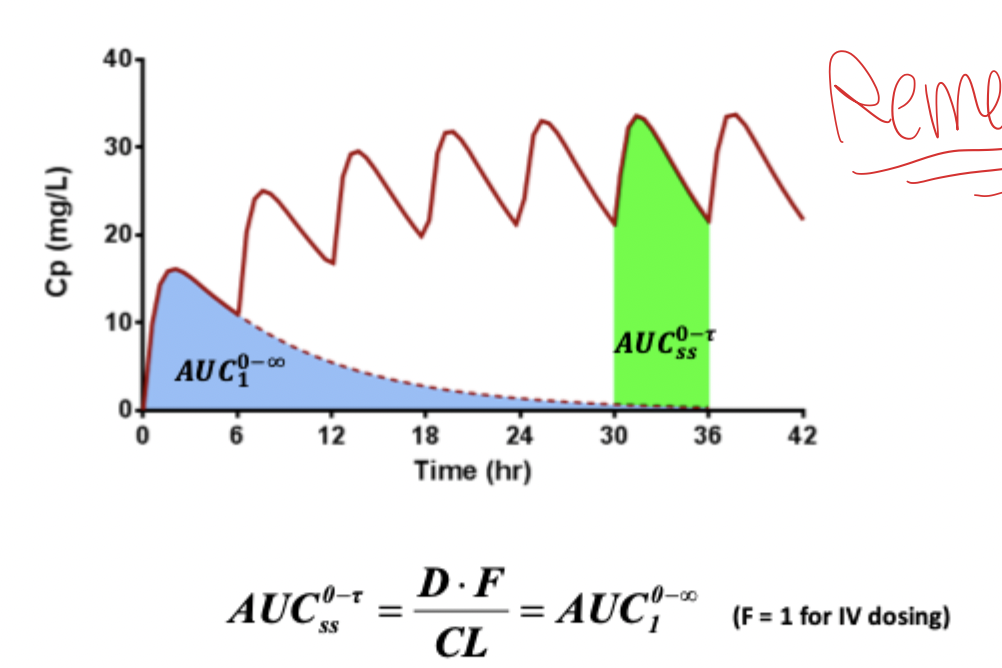
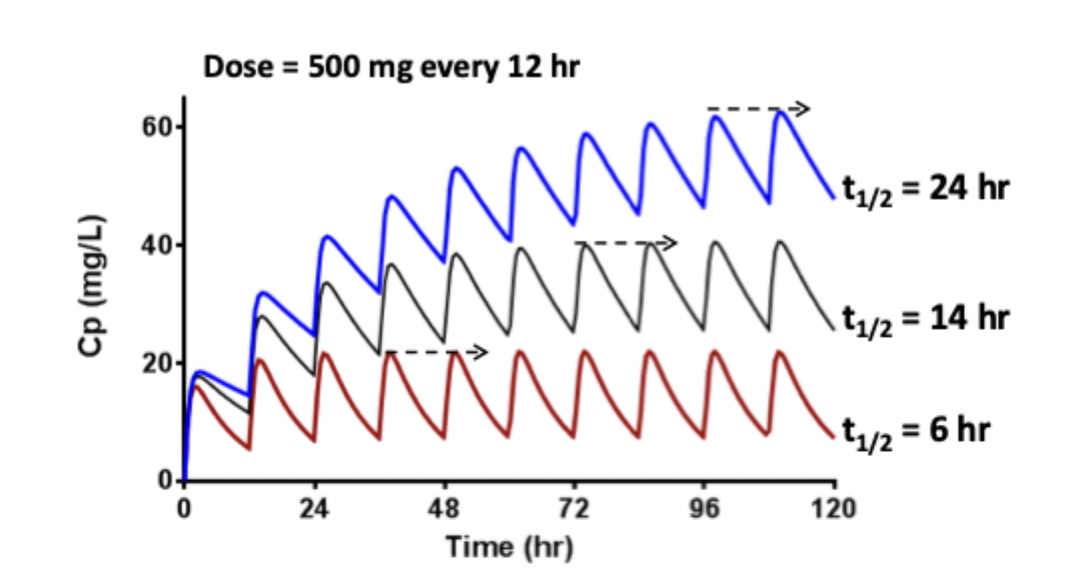
Time to Reach Steady State
Only dependent on elimination half life
Remember (kel = cl / v )
Noyes - Whitney Equation
Parameters affecting dissolution
Diffusion coefficient → molecular size → increased by smaller sizes
surface area → particle size → increased by smaller particles
diffusion layer thickness → increased by faster stirring
Saturated solubility of drug → intrinsic solubility → composition of medium, pH of medium
Drug concentration → increased by sink conditions
Dissolution Testing Apparatus
Apparatus 1 → basket
tablet in basket that rotates vertically
Apparatus 2 → paddle
paddle rotates in beaker
Lipinski’s Rule of 5’s
Good in vivo oral drug absorption and permeation:
logP < 5
H bond donors < 5
Molecular weight < 500
H bond acceptors < 10
Why does LogP matter?
Increasing LogP
increases binding to enzyme & receptor
increases absorption through membrane
decreases aqueous solubility
increases binding to metabolizing enzymes
increases binding to blood proteins
Biopharmaceutical Classification System
I → high solubility high permeability
III → high solubility low permeability
II → low solubility high permeability
IV → low solubility low permeability
highly soluble → in 250 mL or 8 oz. of water
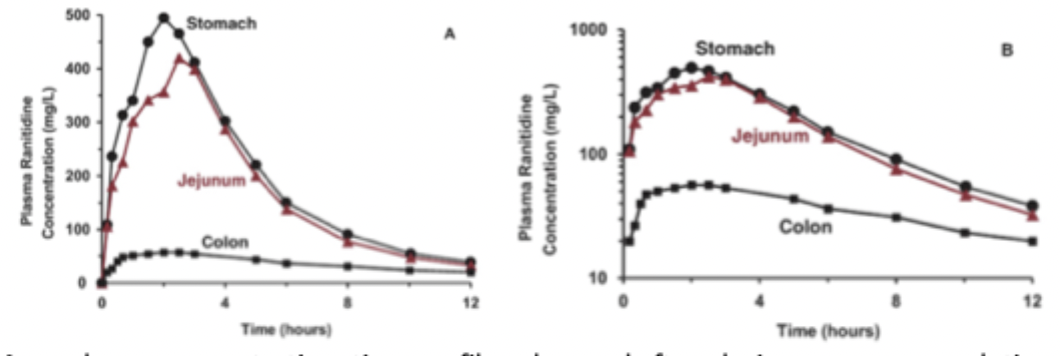
Site of Absorption & Permeability
There can be regional differences in where a drug is absorbed
drugs with very low permeability are likely to be incompletely intestinally absorbed
show low bioavailability because of the limited time in the small intestines where permeability is the highest
Gastric Emptying
Water leaves stomach fast, than digestible solids, and than large undigestible solids
Because absorption is greater in small intestines, rate of gastric emptying can be a controlling step in drug absorption
gastric retention can be utilized as a mechanism for sustained release oral drug absorption from the small intestines
Absorption Window
If absorption is mediated only my uptake transporters in a small region, then drug absorption is limited to that window
drug release must occur before the absorption window
Big dose of drug is not recommended either because the uptake transporters are often saturable
Lag time
For some drugs, absorption does not start immediately, due to delay in gastric emptying time or intestinal motility
Site of Injection
absorption from different areas of the body may differ
the greater the speed of absorption, the more rapid the exposure, the shorter duration of action
Effect of Food
Food may effect both rate and extent of absorption
positively or negatively
FDA requires studies on food effects on all new drugs, especially sustained release products
Large meals can delay gastric transit time
Some medications (ex. antibiotics) should not be taken with dairy products due to chelation to ions
Some medications are more soluble in the stomach, so a big meal delaying gastric emptying will allow more drug to dissolve before entering the intestines, increasing bioavailability
However this could be negative if a sustained release dose stays in stomach for too long ending in dose dumping
Enterohepatic Circulation & Charcoal
Drug is reabsorbed back into circulation
Orally ingested charcoal absorbs drugs in the intestines, preventing their absorption and future enterohepatic circulation
Intrinsic Clearance
Ability of the organ (liver) to remove drug in the absence of flow limitations and binding to cells or proteins in the blood
High and low ER Drugs (IV)
High ER (>0.7) → blood flow limited
Low ER (<3.0) → Fu x Clint limited
Oral Clearance
Fu x Clint = Cloral
NOT dependent on blood flow
High and low ER drugs only influenced by changes in Fu or Clint
Factors Affecting low ER drug PK
IV
Increase Clint → increase in clearance
Increase Q → no change
Oral
Increase Clint → increase in clearance
Increase Q → no change
Factors Affecting high ER drug PK
IV
Increase Clint → no change
Increase Q → increase in clearance
Oral
Increase Clint → increase in clearance
Increase Q → higher Cmax and increase in clearance but same AUC
Brand vs Generic
Same active ingredient
Same strength and dosage
Generic is cheaper, almost always covered by insurance
Inactive ingredients may differ but have to be accepted by FDA
Appearance and look may differ
Generics are cheaper in cost
Bioequivalence
FDA has set standards on rate and extent of absorption by which two products can be considered bioequivalent and therefore able to be substituted for one or another
Extent → Log AUC
Rate → Log Cmax
Adjusted for log transformed data, the range becomes 80% - 125%
Thus the two products are bioequivalent if all 4 characteristics are between 0.8 and 1.25
Bioequivalence Testing Preference
In vivo measurement of active moiety in biological fluids
In vivo PD measurements
In vivo clinical comparison
In vitro comparison
Bioequivalence Calculation
Degrees of freedom = subjects (n) - 1
Confidence = 90% confident → 1-0.9 = 0.1 / 2 = 0.05
mean difference = e ^ MD
Cmax = e ^ Cmax
mean difference and Cmax confidence intervals → use equations. Remember MD is not the same as e^MD value