DAPAB W4L2/W4D3: Mixed model using all relationships and ReML
1/43
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
44 Terms
Main traditional reasons for mixed model
Non-independence/estimate the fixed effects accounting for all the noise (P-values, SE)
Estimating variance components: reasons for ReML
ANOVA does not work for VCE with unbalanced data
Use ReML
Traditional reasons for mixed models and ReML
Recovery of inter-block information with incomplete blocks
Use ranodm block effects for identifiability of fixed effects of interest
In animal breeding, interest is in the
random breeding values → BLUPs
and in the aditive genetic ariance in non-designed populations (dismgaA) → ReML
The fixed effects merely serve as correction factors (herd, year, season age)
The BLUEs of fixed effects are usually not of main interest in animal breeding
Mixed models and ReML in animal breeing
Interest is in the random effects
Breeding values
Gene effects
→ need the variance components
Unbalanced data
unequal family sizes
General pedigrees
Related sires and dsams
Any type of relatedness
Use all family information, e.g. all dutch cows
Non-random samples
selection of superior individuals
Non-ranodm mating
Include genomic informaiton
Genomic breeding values
GWAS
Mixed models and ReML in plant breeding
Interest is often in the fixed effects
Unbalanced data (e..g incomplete blocks)
Include genomic information
GWAS
Fixed or random
Simplicity
Fixed effects are simpler, models converge better (least squared instead of ReML)
Conceptual reasons
Is your model term a random sample of a population?
Sex effect: NO, there are only two sexes→ sex is fixed
Pen: YES, there could be many different pens → pen is random
Avoid false significance in hypothesis testing (avoid too small P-values)
For example: does sex affect body weight of pigs
Include sufficient noise in the model to avoid false significance
Test for sex effect: fit pen, batch etc as random effects
Avoid overstimation of effects (BLUPS)
Random effects (BLUPs) get shrinkage → estimates are conservative
Fixed effects (BLUEs) get no shrinkage → suffer from winners curse
Fixed effects are greedy
Fixed effects absorb too much varaition when classes are small
Partial confounding, fixed effects have priority over random effects
Recovery inter-block information → blocks must be random
Fixed variety effect has priority over the random block effect
So inter=block info can be utilized
Relationships/correlations
Random effects can be correlated (e.g. related animals)
→ genetic effects are treated as random
Real life: why did we not simply skip ANOVA
ANOVA requires many assumptions that never hold
Real data are never balnaced
Sires and dams tend to be related
Preselection is common in breeding populations
ANOVA shows where the information comes from → helps understanding of what we do
ReML is way more powerful, but we do not see where the inforamtion comes from
For balanced designs; ANOVA and ReML are
Equivalent
The general mixed model for genetic analysis

The general mixed model for genetic analysis
Y is a vector → the variance of y is a matrix V

Animal MOdel: the simplest mixed model for genetic analysis
Residual identical distributed

The u-effects (breeding values) of related animals are
Positively correlated, related animals have similar breeding values
About the (co)variances of the genetic effects, var(u)

Relationship matrix
Matrix of relationships between all individuals in the analysis
Pedigree relationship matrix A
Genomic relationship matrix G

The pedigree relationship matrix A
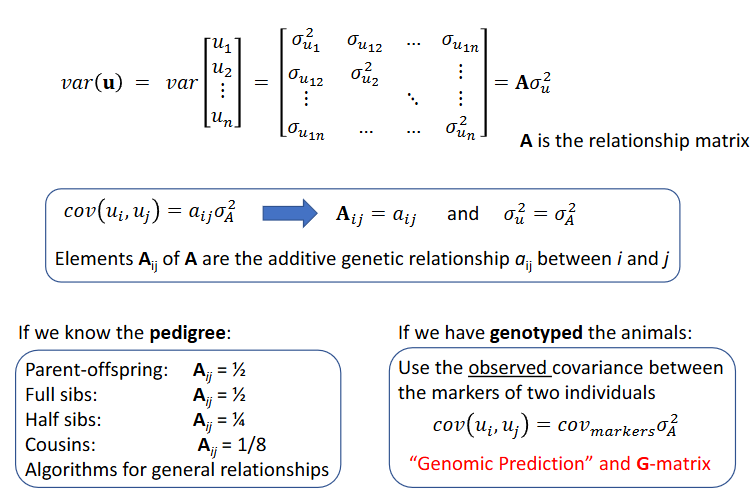
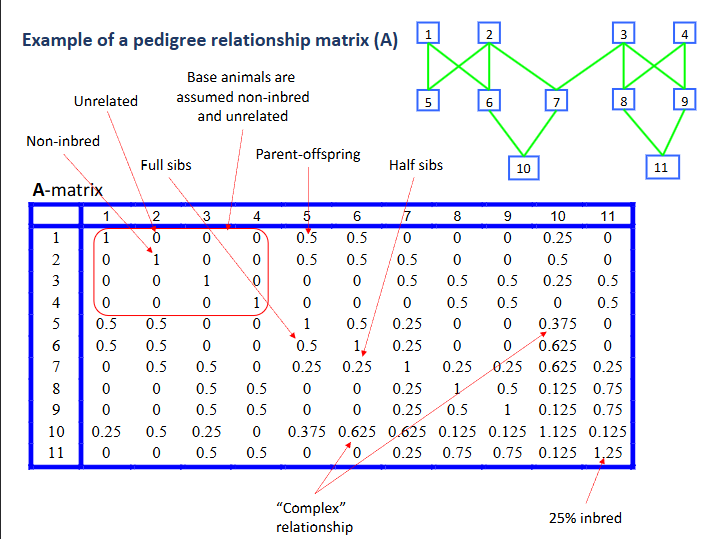
Base generations

Example of a pedigree relationship matrix A
Summary: the simplest mixed model for genetic analysis

Models for special cases
Sire model
Common environmental effects (litter effects)
Repeated records and permanent non-genetic (environmental effects)
The sire model
Sometimes, we don’t know the mother of an individual, while the father is known (group mating and natural birth, sheep etc). The number of animals is very large, the full model becomes too big to fit
Then we may fit the genetic effect of the father (sire) instead of the animal itself

The sire model - interpretation
Interpretaion
similar to the half sib model
- Offspring of the same sire are half sibs → var(sire) = cov(half sibs)
Cov(half-sibs) = 1/4 var(A)
The sire variance is an estimate of a quarter of the additive genetic variance
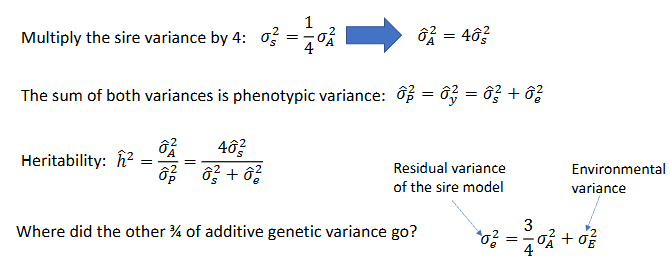
The sire model - assumptions
Without genetypes or pedigree: var(s) = Isigmas2
the sires are assumed unrelated
The sires are assumed to be random sample of the population
The dams
are assumed to be random sample of the population
unrealted to the sires
mated at random to the sires
each dam contributes a single offspring (No full isbs in the data)
Same assumptions as for the HS design with ANOVA (no need for balanced data)
What would happen if we appply a sire model to a pig population?
pig data contain full sibs
We overestimate heritability
Common environmental effects - piglest born in the same litter
Are full sibs (usually)
Developed in the same uterus
Also receive the ordinary animal model to data on weaning weight: y=Xb+Zu +e
Solution for common environmental effects
The common environmental model: y=Xb + Xu +ZCec +e
ec: vector of random effects common to litter mates (common environment)
Zc: matrix linking observations on indiviuduals to their birth litter
Litter effects are assumed independent 𝑣𝑎𝑟 𝐞𝒄 = 𝐈𝜎𝐸2
Genetic effects are correlated via the pedigree or genotypes: var(u) = 𝐀𝜎𝐴2
The common litter effect also captures (most of) the dominance variance, therefore we don’t usually fit a separate dominance effect
We can separate litter and genetics when
The sire is not completely confounded with litter, thus HS provide info for simgaA2
The different dams may be related, but their litter effects are independent
Therefore the ReML likelihood depends on sigmaA2 and we can estiamte it
With FSHS ANOVA we could not separate genetic and non-genetic dam effects
But
The full-sib covariance is completely confounded with litter, thus litter effects reduce precision (and power) to estimate SigmaA2
Repeated records - some traits we may record multiple times on the same animal
Body weight at different ages, milk yield each week or month, litter size in different parities
Suppose we fit the ordinary model to repeated records on body weight
The similarity between repeated records of the same individual (i) is assumed fully genetic
But what if
the animal has been sick when it was young, poor nutrition when it was young → permanent environmental effects (developmental effects)
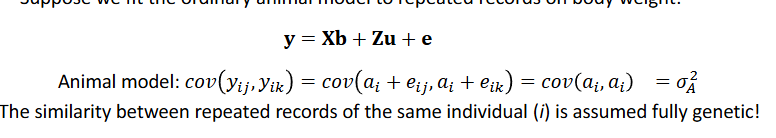
Repeated records - permanent environment model

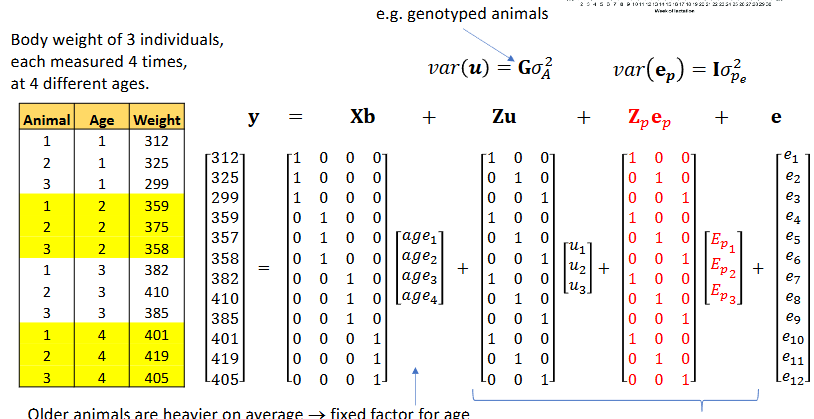
Repeated records - example
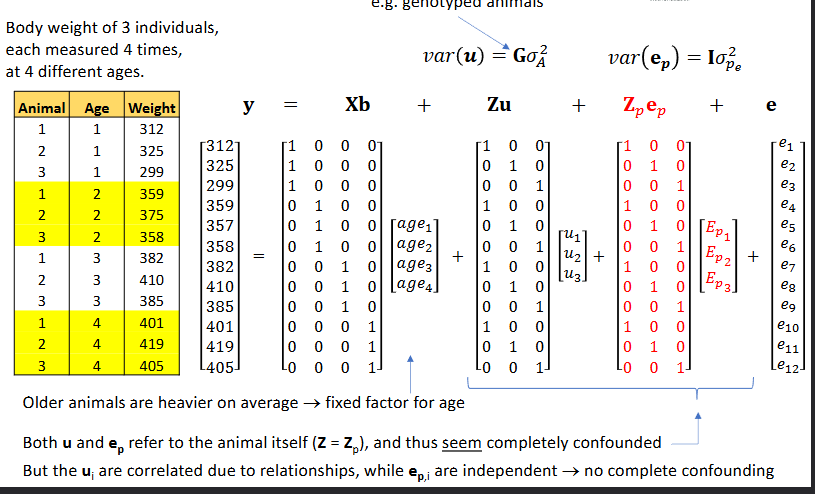
Repeated records and common environment
Data consists of the following:
For each ltiter, we have weaning weight of each piglet
Within a ltiter, piglets experience a common environment (Ec)
But piglets in different litters are also nursed by the same mother (Ep)

Genetic relationships
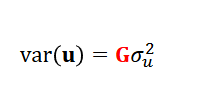
Geneomic relationships
replace the pedigree relationship matrix A with the genomic relationship matrix G
→ more powerful than pedigree, similar to pedigree
Genomic relationships and the G-matrix
If we have genotyped the animals:
Use the observaded covariance between the markers of two individuals
cov(ui,uj) = covmarkersSigmaA2
Genomic prediction and G matrix
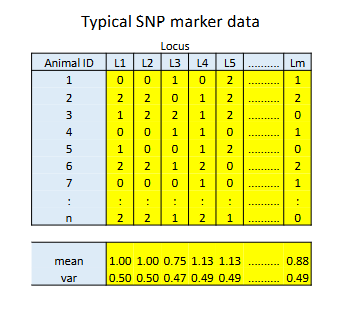
MARKERS = SNP-markers
SNP-markers
m marker loci
Each locus has 2 alleles, e.g. A and T
We count one of the two alleles, say A
Diploids → counts are 0,1 or 2, TT=0, AT and TA are 1, AA=2
p is the frequency of the counted allele - count ~ BIn(n=2,p)
The mean allele count is 2p
The varaince in allele count = 2p(1-p)
Genomic relationships: the observed covariance between markers of animals
The common way to calculate a covariance from a sample
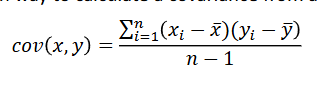
Genomic relationships: the observed covariance between markers of animals
With markers - covariance
We want the covariance between two individuals, say i an j, due to all the m markers
x and y are the allele counts in the two individuals, with values M = { 0,1,2}
For a specific locus (l), the mean x and y is two times the allele frequency, 𝑥(^-) =y(^-)𝑙 = 2𝑝l
There are many loci, so n -1 = n, ignore the -1
Genomic relationships: the observed covariance btween markes of animals
The numerator of the relationship between i and j

Genomic relationships: the observed convarience between markers of animals
Next, we want to write var(u) as: var(u) = G(sigmau2)
where 𝐆𝜎𝑢2 is a covariance matrix
Hence, G is a standardized covariance matrix
We have to standardize (divide) the covariance by the marker variance
The resulting geomic relationship in yellow
Gij is the similarity of the markers of i and j, measured as a standardized covariance
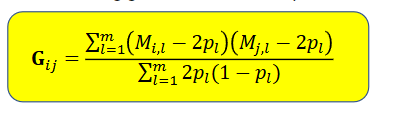
A and G do not depend on the trait, but only on the
Pedigree (A) or on the genotypes (G)
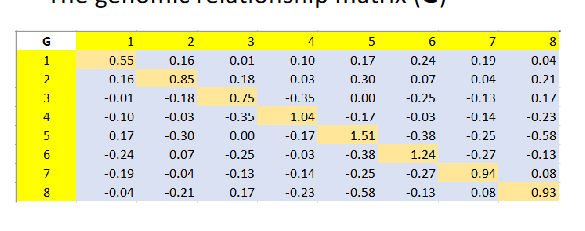
The genomic relationship matrix (G)
Note:
G has both positive and negative values
Off-diagnoals Gij: relationships between different individuals
- positive values → individuals are more similar than average
negative values → individuals are less similar than average
Diagonal elements Gij: self relationships = 1+ inbreeding coefficient
smaller than 1 → individual is less inbred (homozygous) than average
larger than 1 → individuals is more inbred (homozygous) than average
in contrast to the pedigree relatiosnhisp (A), negative relationships and inbreeding can exist
This is because G measures allele sharing, relative to the population aveage
Large sample in HWE: mean off-diagnoals: G(^-)ij = 0, mean diagonal g(^-)ii=1
Base generations
If we fit a genetic LMM, we get an estimate of the addivie genetic variance
BUt to which indivudals or subpopulation does this additive genetic variance refer to
GENOTYPES
In G, we subtract 2p when calculating the covariance → the mean genomic relationship is zero in the pouplation we used to calculate p → with genomic relationships the additive genetic variance refers to the pouplation we used to calculate p, usually htese are the genotyped individuals
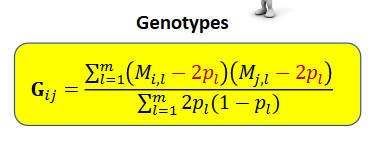
Summary - mixed model using all relationships and ReML
