ceutics xenobiotics across bio membranes (absorption)
1/99
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
100 Terms
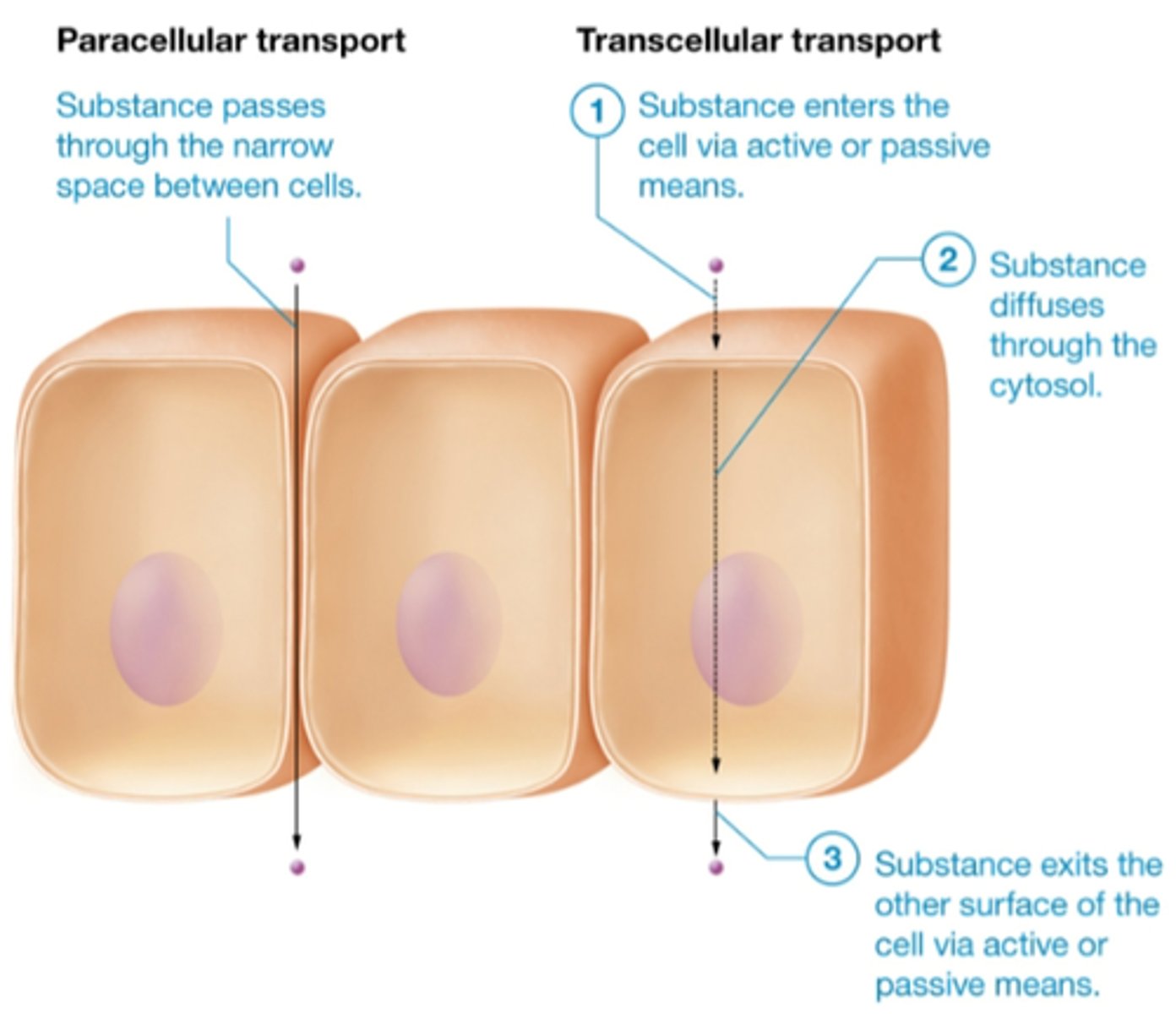
transcellular vs paracellular transport
transcellular (through cell): mostly lipids go straight through
paracellular (between cells): through tight junctions, low molecular weight
why may a drug with paracellular transport be more likely to pass through given a state of inflammation
during inflammation, tight junctions are more loose/permeable, allowing the drug to enter more easily
what does paracellular transport depend on
molecular weight of drug (must fit through tight junctions)
t/f: transcellular transport depends more greatly on molecular weight rather than hydrophobicity
false. more lipophilic drug is more likely to enter transcellularly, even if kinda large
highly ________ drugs tend to diffuse through cell membranes
a. lipoholic
b. hydrophilic
a. lipophilic
t/f: all cells in our body have a cell wall
false, thats bacteria. we have cell membranes.
what can cross the semipermeable cell membrane
water, someee small hydrophilic molecules, lipophilic molecules
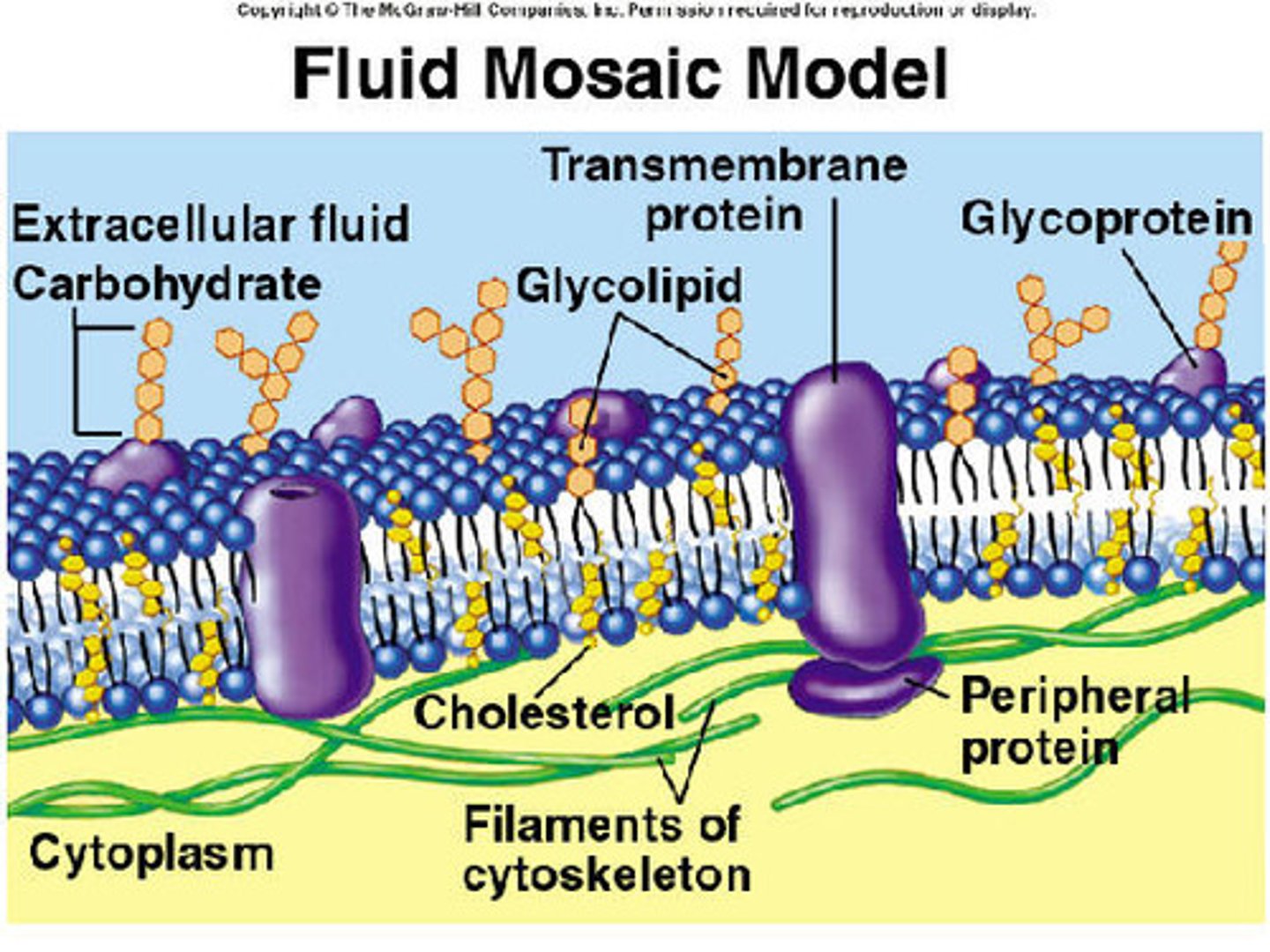
fluid mosaic model
model of cell membrane structure=lipid bilayer matrix with embedded globular proteins
- proteins provide a pathway for transport of small hydrophilic molecules (ion. glucose_ across lipids
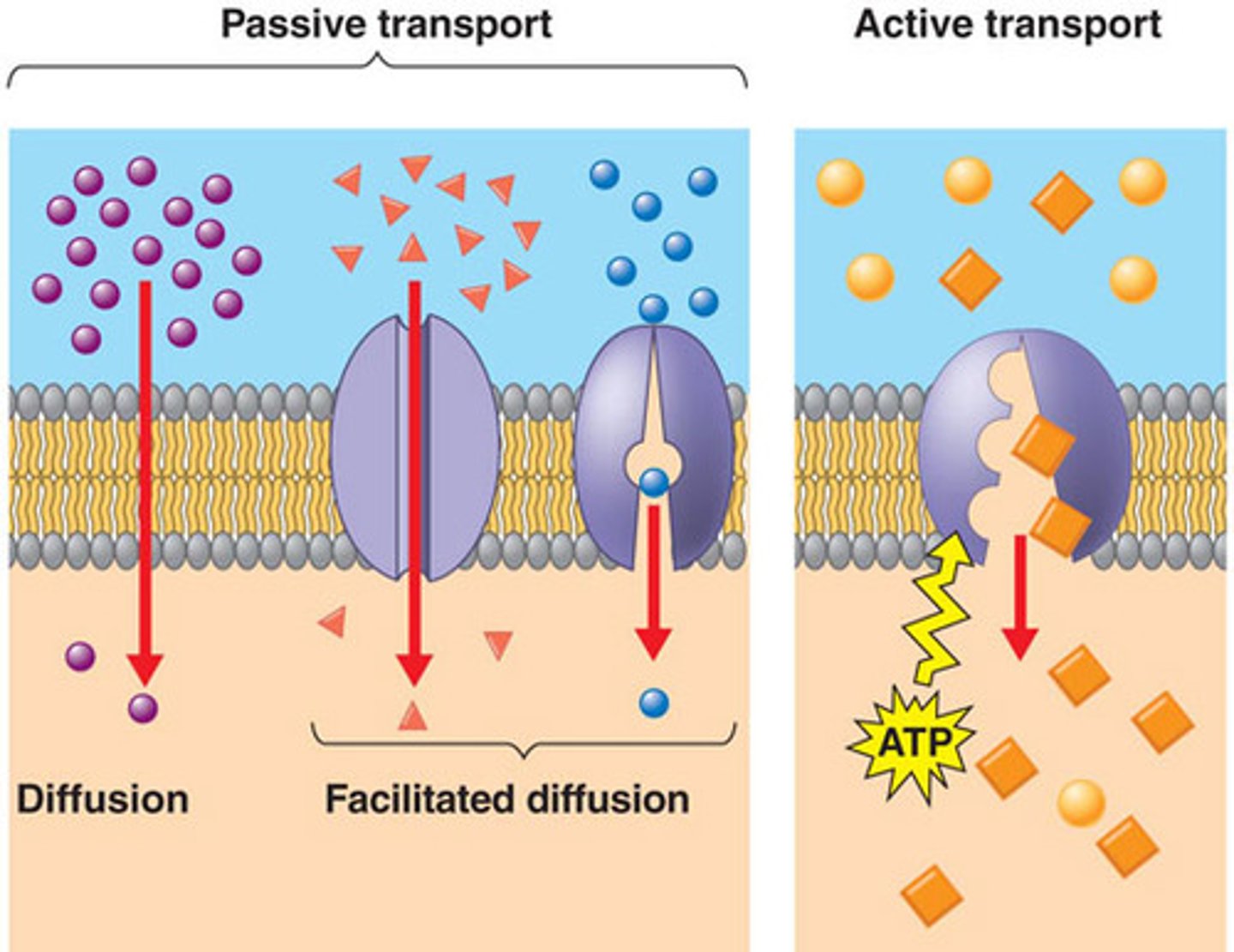
passive diffusion
definition/characteristics
spontaneous diffusion from HIGH to LOW concentration (usually lipophilic drug)
- NO energy required. driven by gradient
t/f: at equilibrium, the number of molecules is the same on both sides of the membrane
false. the rate of movement on both sides is equal, but that doesnt mean the number of molecules is equal (no net transfer)
if equilibrium is reached at the absorption site for a xenobiotic, how can we increase absorption?
increase blood flow to establish a stronger concentration gradient so that drug molecules will move into blood/lymph
given a pt with polycythemia, is the time to equilibrium for a drug going to be greater or lesser
lesser bc blood flow is sluggish so equilibrium is established faster
ficks law of diffusion
(know equation)

t/f: usually, concentration in plasma< concentration at site of absorption
true (takes time to be absorbed into blood)
(this is also why Cp is negligible in ficks law of diffusion)
t/f: lipid molecules follow fick's law of diffusion
false. they just pass through, they dont rlly follow pharmacokinetics
what explains why more drugs are absorbed in the small intestines
small intestines have villi that increase SA
how can Fick's law be manipulated
given:
rate= (DAK)/h (Cgi -Cp)
-D, A,K, and h are all constant so can be coined as constant "P"
-Cp is negligible bc theres way more drug at absorption site than plasma
so becomes
rate= P (Cgi)
where Cgi is [absorption site]
absorption under fick's law is an example of a _____ order process
first order bc absorption is proportional to concentration of drug (not just a constant flow as seen in zero order)
why is absorption usually faster than distribution given fick's law
-drug concentration is higher at site of absorption than in blood so it takes less time for drug to enter blood
-it will take more time for drug to exit blood bc theres a small concentration gradient btwn blood and tissue
(Cgi>>Cp)
most drugs are ______ electrolytes.
what characteristics does this give them?
weak electrolytes
-both hydrophilic and lipophilic
- nature of drug depends on pH/ ionization
________ drug is water soluble while ________ drug is lipid soluble
(ionized or unionized?)
ionized= water soluble
unionized= lipid soluble
given pKa of salicylic acid= 3 and plasma pH=7.4 and stomach pH=1.2, where do you expect higher concentrations?
-in stomach it would be UNionized bc its acid in an acid (HX)
-in plasma it would be IONized bc its acidic drug in basic environment (X-)
- this means that theres very little drug in stomach, it has crossed over into plasma which means its a weak acid
if a drug is not in the stomach and has instead crossed over into plasma due to its charge, what kind of drug can it be
weak acid or ethanol
drug X is a weak base. do you expect more drug in stomach or plasma
a weak base in the stomach would be IONized since it gained H so it would not absorb into plasma. we expect more drug in stomach and very low in plasma
(ion trapped in stomach)
weak ________= more in stomach than plasma
weak _______= more in plasma than stomach
weak bases= more in stomach than plasma
weak acids= more in plasma than stomach
which parameter of pharmacokinetics is related to "binding to plasma or tissue proteins"
distribution
if a drug is highly bound to plasma proteins, how does this affect half life and affinity
longer half life (stay in plasma longer) and lesser affinity to tissue
if a drug is highly bound to tissue proteins, how does this affect half life
short half life bc affinity for tissue will draw it out
why do anesthetics easily enter into CNS and have quick effect
brain tissue is lipophilic and BBB keeps other substances out. lipophilic substances easily pass and stay in adipose
(anesthetics = highly lipophilic)
which drugs are known to complex with calcium and stay in bone/teeth
tetracyclines
tetracycline and calcium interaction
form non-absorbable complex= this lowers absorption and lowers therapeutic effect
which drugs do the following tissues specifically uptake (sequestration)
thyroid=
intracellular water=
adrenergic storage sites=
thyroid= iodine
intracellular water= potassium
adrenergic storage sites= catecholamines
molecules greater than ______ Da require specialized carriers to cross membranes
600-1000Da
t/f: active transport can be seen in both the site of absorption and sites of elimination
true (GI, renal, biliary secretion)
t/f: active transport is only used to move molecules against their concentration gradient
false. its used for both down and along the gradient. active just means it requires energy
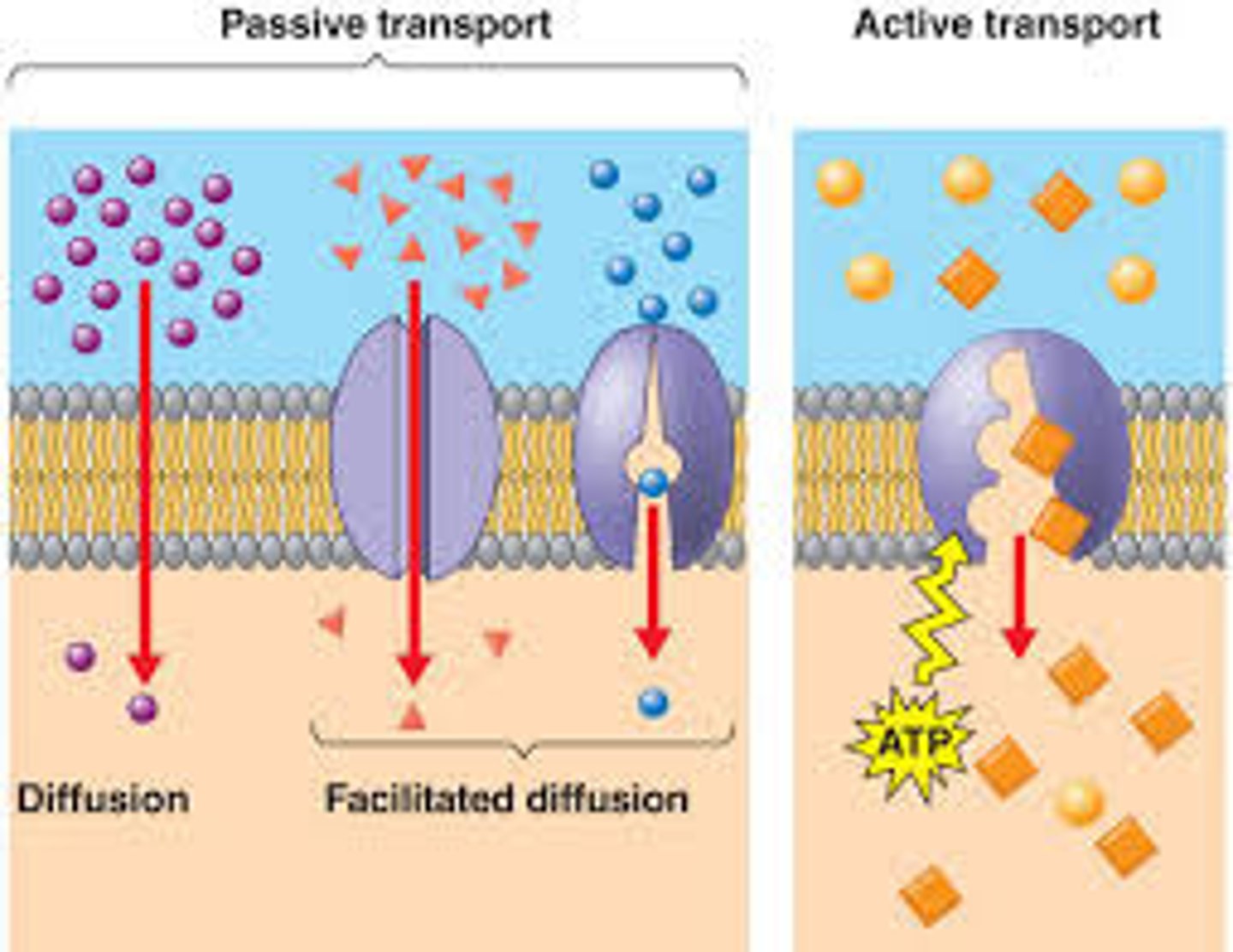
2 types of carrier-mediated transport
1. active= ENERGY REQUIRED
2. facilitated= NO energy required
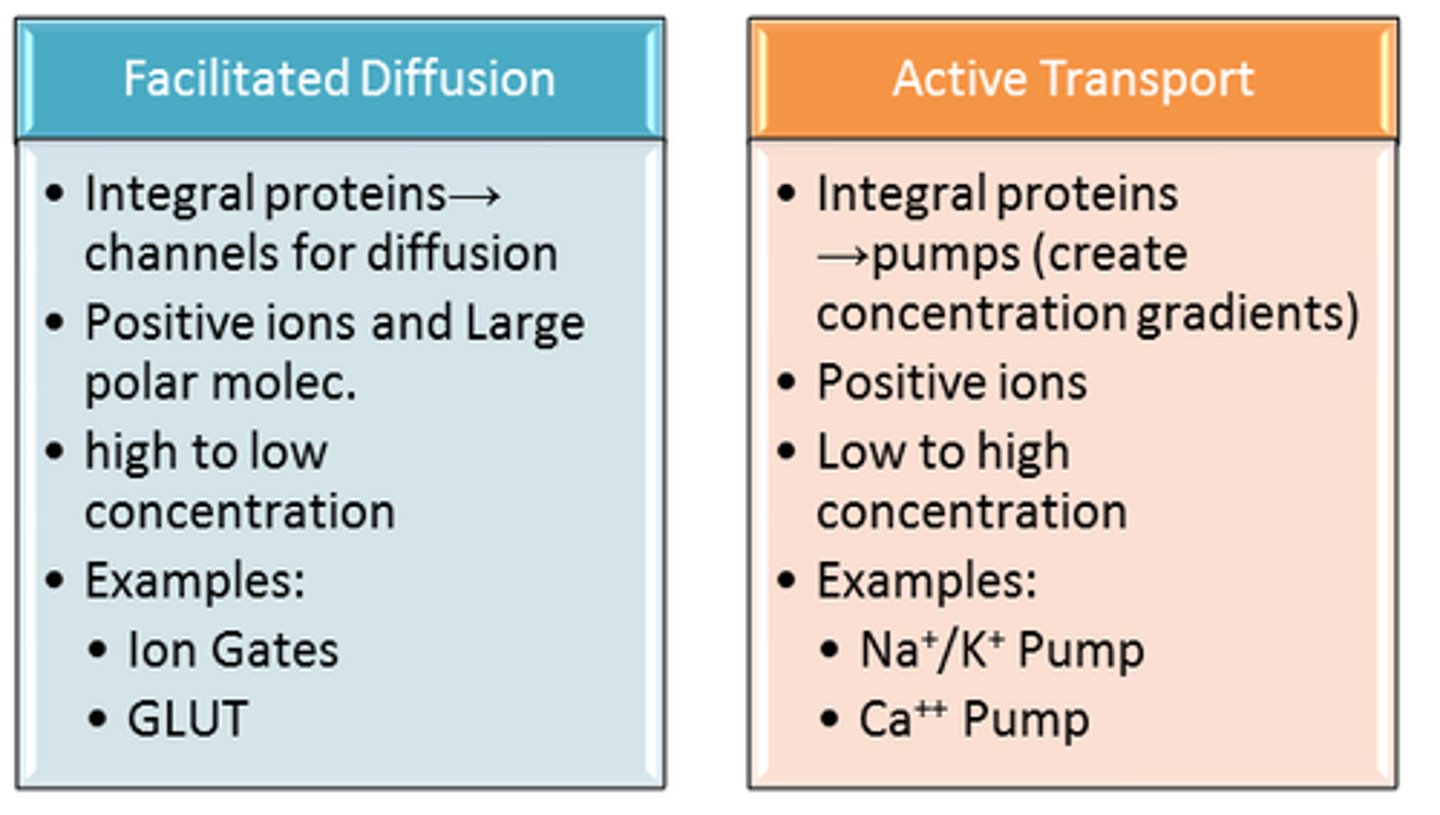
active transport
1. requires energy
2. down or along concentration gradient
3. requires specific carrier that will form complex w drug
describe competition of drugs in active transport
-if a drug is using a specific carrier its competing with endogenous substances and other drugs with similar structures
-if a bunch of drug molecules are competing for same carrier then overall transport is slower unless if 1 drug has more affinity than others
describe saturation in active drug transport and compare it to diffusion
-doesnt matter how many high affinity drugs there are, carriers only let in certain amount per time (gradient does not drive rate!)
- in diffusion the stronger the concentration gradient the greater the rate (gradient drives rate!)
t/f: in active transport, the gradient drives the rate
false. saturation and energy drive rate
t/f: carrier mediated transport is saturable
true. rate of transport does not increase beyond a certain concentration
the highest density of transporters is found in
first 2-3 meters after stomach (proximal jejunum)
p-glycoprotein (P-gp)
where is it on cell?
what does it do?
effect on ct curve?
-found on apical cell membrane of GI tract (facing gut contents, not capillaries)
-drug molecule enters gut and begins to go through cell and towards capillaries. p-gp pumps this back onto inside of the gut so that it doesnt reach capillary
effect on ct curve: shifts curve down (bc less absorption since its not reaching capillary)
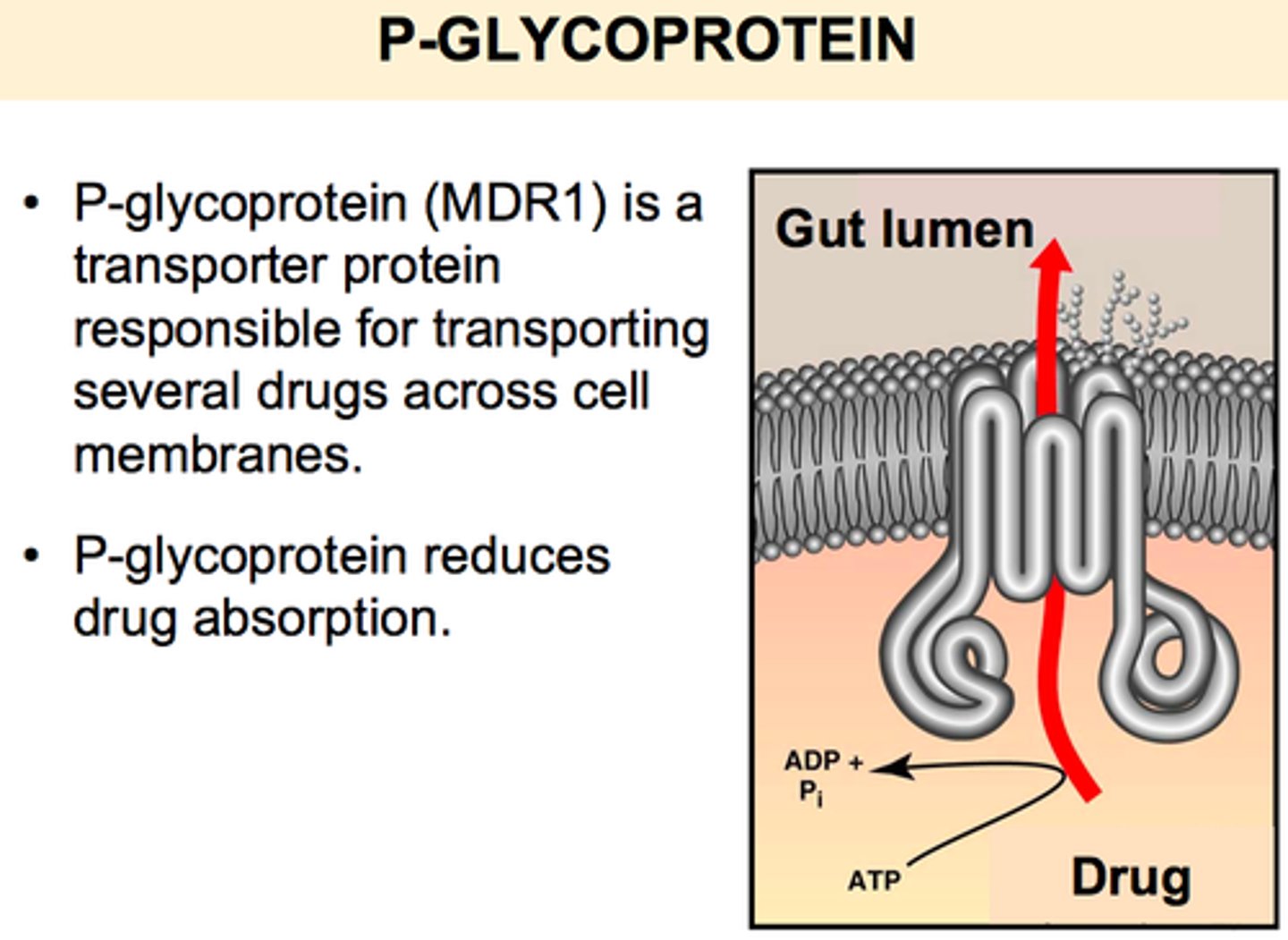
how will p-gp shift the concentration time curve
down (bc less absorption/ less drugs reach blood)
function of p-gp based on location
intestine:
liver:
kidney:
brain:
intestine: prevent absorption
liver: excretion into bile
kidney: excretion into urine
brain: prevent entry from blood into brain
t/f: p-glycoprotein is an example of carrier mediated protein used in facilitated diffusion
false. active transport bc requires ATP (energy)
drug X undergoes significant p-gp activity. what is the effect on drug intensity?
decreased intensity bc does not reach blood so lower concentration of drug in blood and site of target
a formulation of a drug notorious for p-gp would be made to have _____ potency
higher potency bc less is expected to reach blood due to p-gp
2 ways p-gp is induced
1. increase amount of p-gp (more pgp in general)
2. increase intrinsic activity of the same # of p-gp (same amount of pgp will process more substrates)
effect of inducing p-gp on ct curve
will shift curve even lower bc more pgp= less reaching blood= less absorption
effect of inhibiting p-gp on ct curve
less pgp means more drug is reaching blood= higher absorption= shift up
which drug can induce p-gp? how/effect?
- rifampicin increases amount of p-gp
- reduces absorption of other substances
a drug that induces p-gp will lead to _________ of other drugs
a. less absorption
b. more absorption
c. greater duration
d. shorter duration
a. less absorption
- more pgp means more drugs are pumped back into lumen and less reach capillaries
which drug can inhibit p-gp? how/effect?
- verapamil will saturate p-gp, not allowing other drugs to bind
- increases absorption of other molecules since pgp is being occupied by the verapamil
a drug that inhibits p-gp will lead to _________ of other drugs
a. less absorption
b. more absorption
c. greater duration
d. shorter duration
b. more absorption
- if theres less pgp that means less drugs are being pumped back into lumen and more drugs are reaching blood
facilitated diffusion
- no energy required but does require transporter
- saturation and competition
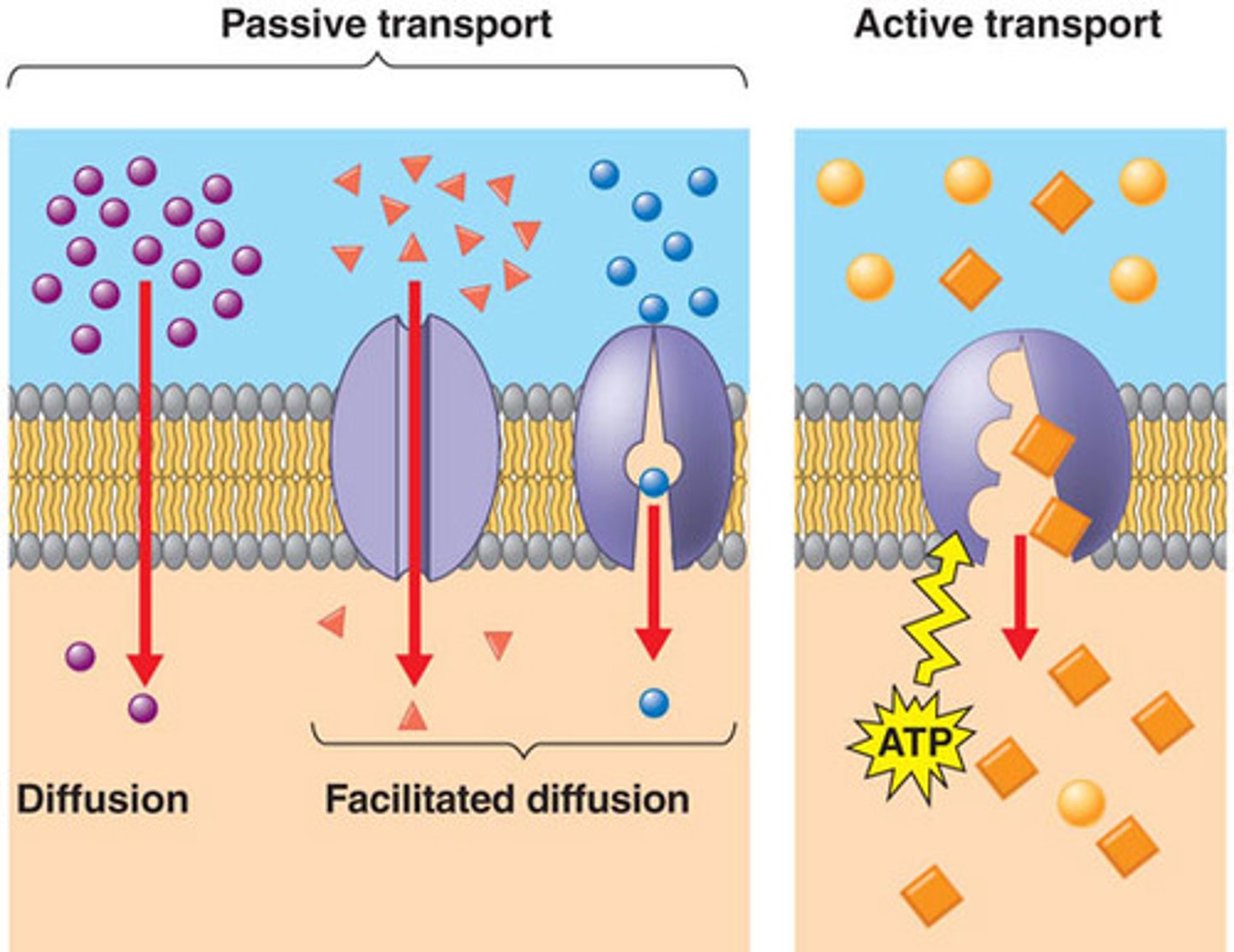
diffusion vs facilitated diffusion
diffusion: rate is dependant on strength of concentration gradient
facilitated: rate is dependant on carrier saturation, not just gradient. transport only moves select amount per time
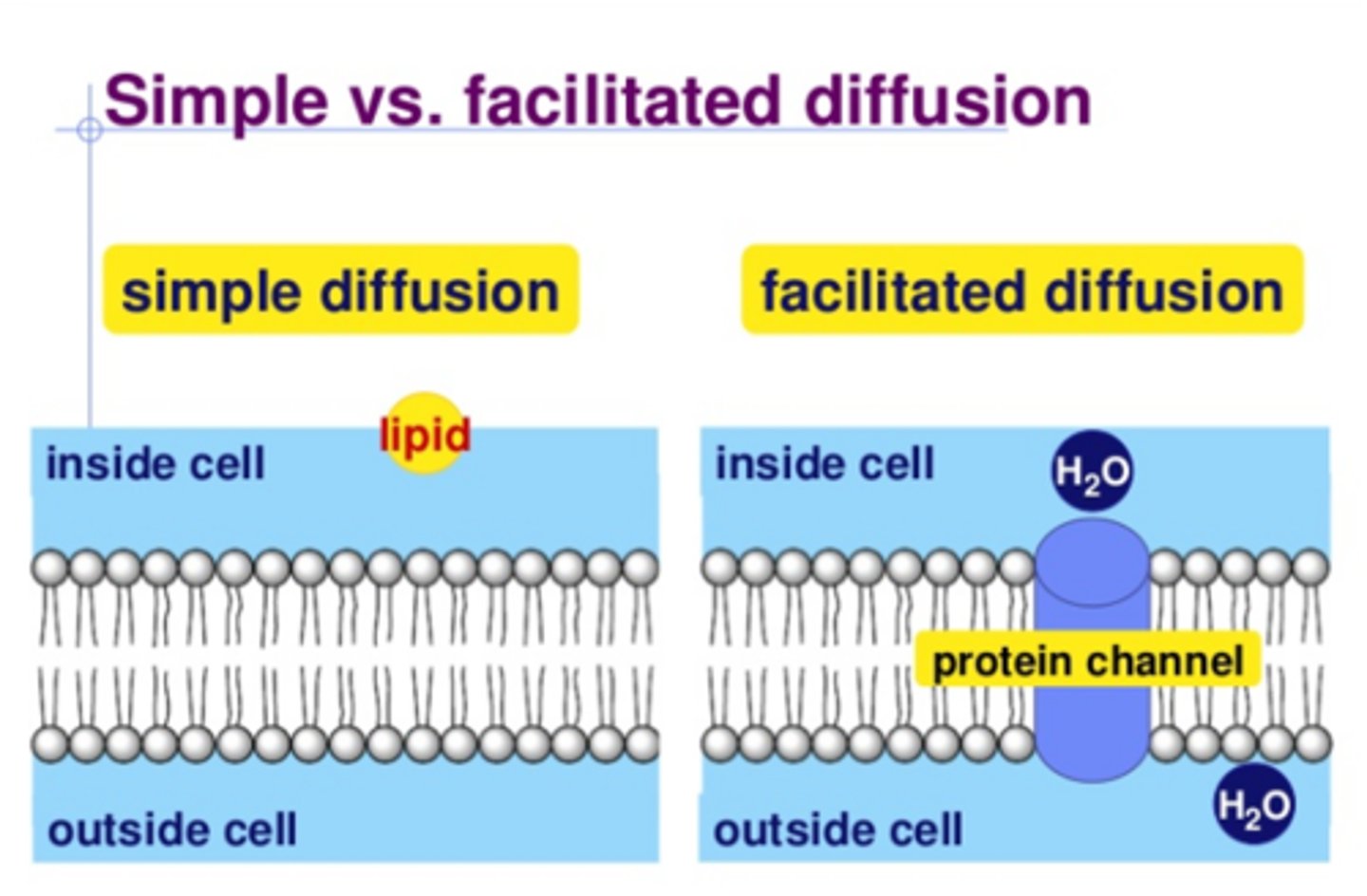
t/f: increasing the concentration of a drug undergoing facilitated diffusion will increase rate of absorption
false. facilitated diffusion has carrier protein that is saturable and has constant rate. increasing drug will not increase rate [unlike in simple diffusion which WOULD increase rate]
![<p>false. facilitated diffusion has carrier protein that is saturable and has constant rate. increasing drug will not increase rate [unlike in simple diffusion which WOULD increase rate]</p>](https://knowt-user-attachments.s3.amazonaws.com/8b4ebbce-e7ec-4392-aca8-da0a84a92217.jpg)
t/f: transport mechanisms that require carrier proteins (such as facilitated diffusion and active transport) are subject to competition and saturation
true
which forms of transport are concentration gradient dependant? which are not/have constant movement?
gradient dependant: passive diffusion
independent/constant: facilitated and active
[saturation and competition]
endocytosis
definition?
2 types?
- process of moving substances into cell
pinocytosis: engulfing liquids and small things
phagocytosis: engulfing large things or insoluble substances by macrophages
exocytosis
release of substances out a cell by the fusion of a vesicle with the membrane
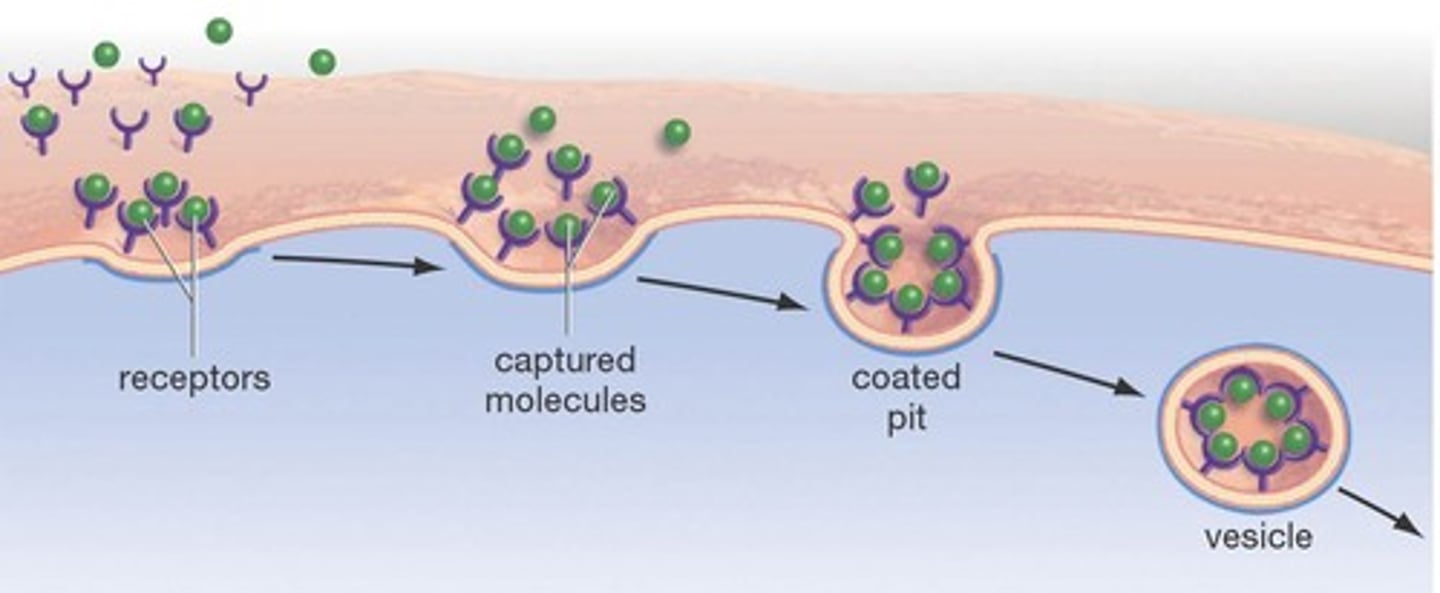
receptor-mediated endocytosis
binding of ligands to receptors triggers vesicle formation that moves thing inside cell
pore transport
what can cross membrane? how?
- small molecules (glucose, urea, water) rapidly cross lipid membrane through nanometer pores
-transport proteins (holes) in lipid membrane can let them in
ion pair formation and passage through membrane
- charged molecules cant pass lipid membrane so + and - join to form a pair, or maybe even attach to protein
- this pair has neutral charge and can diffuse across membrane
effect of highly charged molecules on ct curve
highly charged molecules have slower absorption rate= slope of curve is more downwards/ less steep.
-and elimination is still constant bc depends on filtration/metabolism, not charge
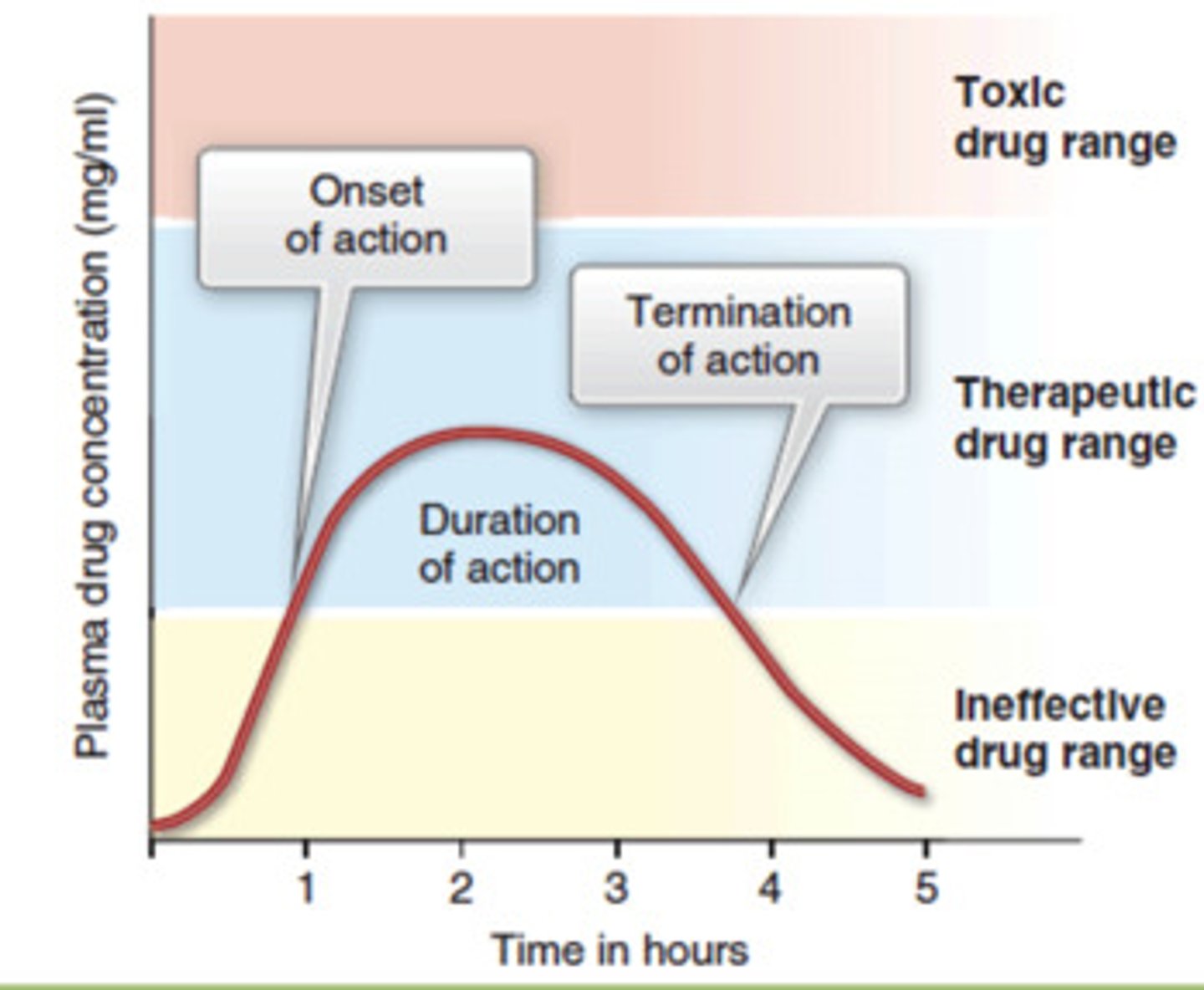
t/f: onset/rapidity of drug is related to its therapeutic activity
false. its time to MEC, not necessarily full therapeutic concentration
most drugs are
a. weak acids
b. strong bases
c. weak bases
d. strong acids
c. weak bases
(absorbed in SI)
t/f: the oral cavity has very low vasculature leading to slow onset
false. oral cavity is highly vascularized, thin lining, AND bypasses first pass effect
ex: sublingual nitroglycerin= highly absorbed and quick onset
describe absorption in the oral cavity
- small SA but very vascularized and thin epithelial lining
- goes straight into blood, bypassing liver and first pass effect
- dissolved drug must be in contact w mucosa to absorb (form and time dependant)
if a pt swallows a sublingual tablet, how does this affect the ct curve?
down
sublingual tablet means it has to be absorbed in the oral cavity so if its swallowed, drug will most likely be destroyed= lower absorption= shift down
t/f: absorption of drugs from the oral cavity detours around the first pass effect
true. it will eventually reach the liver in second cycle tho
buccal/sublingual tabs advantages
- lipid soluble drugs rapidly absorbed (nitroglycerin)
-no first pass effect (straight to blood)
- mouth= neutral ph= good for absorbing drugs (most are weak bases)
which types of drugs are most readily absorbed in mouth
a. weak bases
b. weak acids
c. strong acids
d. strong bases
a. weak bases
absorption site has neutral pH
buccal/sublingual tabs disadvantages
- inconvenience/ taste
- some drug may be swallowed/washed out into stomach
- not for some drugs: polar, high molecular weight, high dose
which kind of drugs are not suitable for buccal/sublingual administration
- polar
- high molecular weight
- high dose
if a buccal/sublingual tablet is pulverized, how does that affect ct curve?
greater SA= shift left bc faster absorption. height does not change bc will only absorb to max amount [unless if theres p glycoprotein]
every drug is assumed to be absorbed to its maximum except if what condition is in place
p glycoprotein
t/f: the primary function of the stomach is NOT absorption bc of limited blood supply
false. the primary function is not absorption but not bc of blood. stomach is highly vascularized
drug X is a weak base. pt has shorter gastric emptying time. how will that affect ct curve?
weak base= not absorbed in stomach
shorter gastric emptying time= moves out of stomach quickly (faster onset)
shifts curve left
drug X is a weak acid. pt has increased gastric emptying rate. how will that affect ct curve?
weak acid= absorbed in stomach
increased emptying rate= leaves stomach quickly/ less absorbed
shifts curve down
drug X is a weak base. pt has lower gastric emptying rate. how will that affect ct curve?
- less of drug is moved into intestines= slower onset
shift curve right
how will the following shift the ct curve
-quicker onset:
-delayed onset:
-greater absorption:
-lower absorption:
-quicker onset: shift left
-delayed onset: shift right
-greater absorption: shift up
-lower absorption: shift down
the stomach may "trap" drugs that are ____________, leading to drug accumulation. this is known as ________
weak bases; ion trapping
where will weak acids be ion trapped
small intestines
(weak bases ion trapped in stomach)
while the SI has all forms of drug transport, the predominant process is _______
diffusion
where does most of absorption in the SI occur? why?
first 1-2m of SI, proximal jejunum
1. greatest concentration gradient since drugs dumped from stomach
2. highest density of receptors
if a weak base spends more time in the stomach, this will shift the ct curve ______
right (longer onset)
absorption in the large intestine
- LI has smaller SA, last line of absorption
- LI has solid contents that impede drugs from touching mucosa and getting absorbed
if there is more solid content in the LI which impedes drugs from reaching epithelial cells for absorption, this shifts the ct curve _______
down
t/f: extremely lipid-soluble drugs enter lymphatics and tissues rapidly, sometimes not even needing absorption into blood
true. may enter lymph right away and avoid first pass effect
t/f: all drugs that are absorbed from the intestines enter the hepatic portal vein and pass through the liver before being distributed systemically. some may be completely removed due to metabolism (first pass effect)
true
if drugs such as mepriride, morphine, aspirin, lidocaine, nitroglycerin, and propranolol undergo extensive first pass metabolism, why/how are they still given
still given but required dose is much greater than for other routes of administration
if a pt vomits after ingesting a drug orally, how does this affect ct curve
less drug= lower concentration= shift down
if the vascular compartment is expanded, what effect does this have on drugs
relative dilution of drugs= lowers concentration gradient leading to less absorption
3 hemorrhoidal veins and their fxn
middle and inferior= good for absorption, bypass liver metabolism by going straight to vena cava
upper= slower and goes to liver
which factors can decrease gastric emptying rate (more time in stomach)?
physiological:
pharm:
physio: solids, acids, fats
pharm: SYMP-> anticholinergics, gang blockers, narcotics/opioids
(think of opioids causing constipation bc slows GI)
for most rapid digestion, we need ________ ANS dominance. this will lead to ______ gastric emptying rate
drug examples:
parasymp; increased gastric emptying
- cholinergics
- anticholinesterases
which factors can increase gastric emptying rate (less time in stomach)?
physiological:
pharm:
physio: liquids, gastric distention
pharm: PARA-> anticholinesterases, guanethidine, cholinergics
how is gastric emptying rate affected based on what you eat
decreased emptying rate: fats, solids, acids (think of heavy meal staying in ur stomach)
increased rate: liquids, distention