TB5: MD
1/185
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
186 Terms
What are the goals of molecular dynamics?
Understaning structure-dynamics-fucntion relationships
static structures cannot provide much information about function without dynamics
Predicting protein-lipid interactions
can be difficult to get with structural data, as proteins have to be individually isolated
Predicting the impact of mutations
Drug design
Identification of druggable binding sites
need to know where this is to determine the size of druggable pockets, and this can be used to design drugs
why may we choose to use computational methods
cleaner and safer
greener? - often requires more electricity and heat
Difficult experimental conditions are easier to achieved
eg. higher temperatures
eg. non-physiologival pHs
Can do unphysical experiments for greater insights
eg. computational can see if interactions are driven by electrostatics - eg. turn off the charged and see if driven by VDWs or not
what does MD use? what does it not use?
uses particles
atomistic - each atom
we do not have electrons !! - we cannot see the formation of covalent bonds
these require new orbitals being formed
we can see H bonds and salt bridges being formed
can see conformaitonal change
what is molecular mechanics
method by which molecular systems are modelled using classical mechanics
what are force fields?
a sum of bonded and non-bonded terms used to describe molecules
what do force fields consider? how can these change?
Forcefields allow considerations of covalent and non-covalent interactions
the covalent interactions cannot be made or broken
but they can expand and contract - known via IR spectroscopy
this is because we do not have representations of electrons
what is the total potential energy of a molecule
the potential energy of a molecule is a sum of the contributions from all of these terms for all atoms
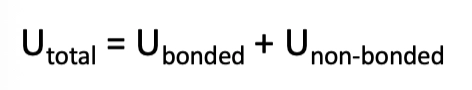
what are the bonded terms in MD?
bond lengths
bond angles
torsion angles
what are the non bonded terms in MD
VDWs and electrostatics
what do we use to represent bond lengths and angles?
harmonic potentials
what to harmonic potentials represent
represent the potential energy for the range of bond lengths/angles
energy on y axis, bond lenght/angle on x
for a bond length harmonic potential., what is at the minima?
the optimal bond length
potential energy equation for bond lengths?
where kb is the force constant, r0 is the equilibrium bond length and rij is the measured bond length
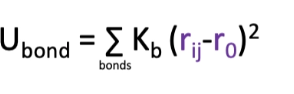
potential energy equation for bond angles?
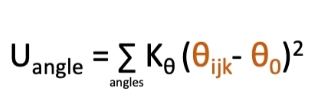
what is the force constant?
the force constant k is the parameter that measures how stiff the potential is.
Large kk → a very stiff potential, stronger restoring force for a given displacement
Small kk → a softer potential, weaker restoring force
why are harmonic potentials not appropriate for dihedral angles?
this is because there are multiple ideal torsions
energies for different configurations can be the same
this means we need a periodic function
what do we use when determining the energy of dihedrals?
We instead use a periodic cosine function
these are defined by 4 atoms and 3 bonds
equation for the energy of the dihedral?
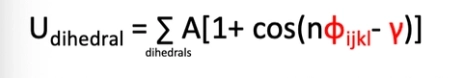
what do the different parameters mean in the equation for a dihedral
A is the force constant, n is the multiplicity, ɸijkl is the dihedral angle and y is the phase factor
this determines the locations of minima
how do we determine the potential energy from van Der Waals
VDWs are modelled via the leonard jones potential
what are the two regimes in the Leonard jones potential? what do they mean
o/r^12 = repulsive regime, o/r^6 = attractive regime
if they get too close, they repel
attraction is the greatest at the minima
as we move away, attraction diminishes
what is sigma on the Leonard jones graph?
sigma is the point where the graph crosses the x axis - this indicates the size of the atom
if they get closer, potential energy goes up - potential energy goes up too much
what is epsilon on the Leonard jones graph?
epsilon, -epsilon is the well depth/stickiness parameter
if epsilon is brought up, it would be easier for them energetically to move away from eachother as they are less sticky
lower -epsilon → more incentive energetically for the particles to stay together
how can we model permanent electrostatic interactions?
we use coloumbs law - accounts for the different charges present, the distance between them
we do this for all the different pairs of charges in the molecule and sum them together
equation for energy of electrostatics?
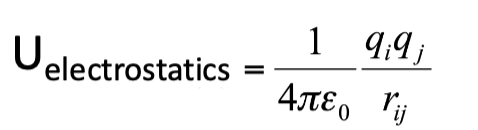
why can electrostatics become a problem for forcefields?
these are long range - 1/r
even when atoms move far away, they will still see eachother, so cannot be observed
this means they need to be continuously calculate
this is the most time consuming part of the simulation
can be sped up
so what does the overall potential energy in a standard force field account for?
bond lengths and angles, dihedrals, leonard jones and the electrostatics
what are the limitations of standard force fields?
Inherently approximate
Lack of polarisation
no electrons, we are assuming that atomic charges are static
really, partial charges on atoms will change, standard force fields fail to recapitulate this
Covalent bonds cannot be made or broken
difficult to look at enzymatic processes
Can be difficult for RNA, IDPs and some sugars
this is due to the lack of structural data, and so we cant get the numbers for the calculations
What experimental data are used to parameterise force fields?
IR, X-ray structures, lipid phase behaviour, NMR exchange rates, partitioning data
How are QM calculations used in force-field parameterisation?
They calculate electrostatic potential, which is then used to fit partial charges.
How are force fields parameterised?
By fitting parameters to experimental data and high-level QM calculations.
how can the resolution of force fields vary?
Can have low and high resolution force fieds
what are the types of force fields we can use?
all atom
coarse grained at the united atom level
coarse grained at the martini level
describe all atom force fields
all atoms are represented
describe coarse-grained united atom level force fields
we take the non-polar aliphatic Hs and combine with the carbons they are associated with
polar ones are left alone - these do interesting thing
we ignore the Hs with the carbons (make a bit heavier/ bigger) to minimise the computational waste
nonpolar aliphatic hydrogens combined with the associated carbon into a single particle
describe coarse-grained martini level force fields
groups of 4 or 5 heavy atoms and their associated hydrogens combined into a single particle
really make the system smaller
but we need to be really careful to make sure we dont use interesting chemistry
eg. we need to make sure different charges are not in the same group, as these dipoles can be very important for the chemistry we are observing
eg. polar and hydrophobic regions cannot be mixed
how many carbons per particle for united atom force fields?
you can only have one carbon per particle
this can only be ch, ch2 or ch3
what do we need to do before MD? how do we do this?
we need to make sure the system is at an energy minimum
cannot be in a strained configuration - the calculations will fail
everything needs to be as close to the ideal values as possible
we use energy minimisation to do this
why is energy minimisation complicated?
lots of different things - bond lengths, angles, torsions etc need to be simultaneously minimise
how do we do steepest descents for energy minimisation?
We want to find the lowest stationary point relative to where we are
do this when the differential is 0
we take the negative gradient of the function to go downhill
how do we go downhill in steepest descents?
we can do this using line searches and arbitrary steps
arbitrary steps - empirical movement backwards and forwards, not very efficient
line search - we fit a quadratic function, solve it to get closer and closer in a more efficient way
what is the problem with steepest descents? how can we overcome?
Problem - steepest descents are good at going quicks downhill, but every direction is a right angle, so you end up zig zagging around the minima
we use steepest descents to get very close to the minima and then switch to another method
eg., conjugate gradients
→ we use multiple methods of minimisation - this is because some vary in efficiency, depending on where they are in relation to the minima
what is the outcome of energy minimisation?
this produces a static set of coordinates
what does statistical mechanics allow us to do>
connect the microscopic and macroscopic world
this allows us to get thermodynamics quantities from MD
what is the ergodic hypothesis?
over a long time period the microstates sampled by MD will match the microstates of the statistical ensemble; so we can take the average of a property over the time of the MD trajectory as the true ensemble average.
what does the ergodic hypothesis mean?
so if we run the MD simulation for long enough, you will het the same properties as the real world
this means we can get an average, but have to make sure that we give the siumulation long enough
what are the potential types of statistical ensembles we can have?
microcanonical
canonical
isothermal-isobaric
what is a micro canonical statistic ensemble?
fixed number of particules, fixed volume, fixed energy
what is a canonical statistic ensemble?
fixed particles, volume and temp
we choose the temp to run at
what is an isothermal, isobaric statistical ensemble?
fixed pressure, temperature and the number of particles
volume can change
what is the input for molecular dynamics>
Input is the suitable initial positions of atoms after minimisaton and the choice of forcefield, along with simulation parameters
what are the simulation parameters we have to choose?
temperature, pressure, simulation length etc
what does the computer do with the MD inputs?
integrates newtons equations of motions and each time it does this, it produces coordinates
what does the computer produce at the end of an MD?
at the end, we get a ‘collection’ of snapshots showing the time evolution of the system
all are related in time
we put these snapshots together to produce a movie that shows how the molecules move with respect to eachother
what are the two equations of motions used?
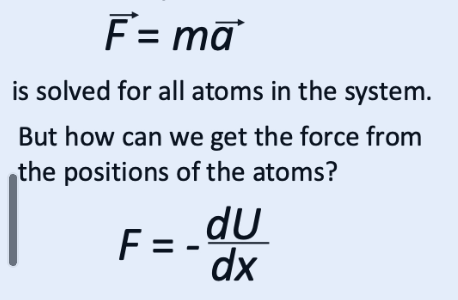
what does the force =-dU/dx equation do?
this produces a direct link between waht the force field tells us and the energy
what is the multiibody problem?
if we want to integrate F = ma, we cannot do this in isolation
each atom will have an affect on the rest of the atoms
we cant solve each in isolation, the moelcules have lnock on effects
→ this becomes very difficult mathematically
so instead, we need to find an approximation method
how do we solve the multibody problem?
We use integration algorithms, these are approximate methods
How does the leapfrog integration algorithm work in molecular dynamics?
Leapfrog is an approximate numerical integration method that breaks motion into very small time steps, δt. At each step, the positions at time t are used to calculate the forces on every atom using the force field, then F = ma gives the acceleration. That acceleration is combined with the velocities at half a time step to update the velocities to the next half-step. Those updated velocities are then used to move the atoms to their new positions one full time step later.
The key idea is that positions and velocities are never calculated at exactly the same time: positions are known at integer time steps, while velocities are known at half steps.
how do we choose the integration time step?
the time step needs to be one order of magnitude smaller than the fastest timescale in the simulation
in an atomistic simulation, this is any bond vibration involving a hydrogen
what happens if we use an integration timestep that is too big?
if steps are too big, this means you could miss some areas, and so wouldn't sample it
what wold happen if integration time steps were very small?
most accurate way would be taking very small steps - bit this is inefficient, it takes ages
what do we balance when selecting integration time steps?
ind a balance between what is efficient and accurate
what could we modify to allow us to use larger integration time steps?
you could fix all bonds involving hydrogens to their equilibrium value, this allows you to use a longer time step of 2fs
this wont change their H bonding
or, you could make every bond angle and length rigid - molecules can translate but there are no internal CCs. this allows you to use much longer timesteps
what is the typical integration time step we use for an all atom simulation?
if all bonds are vibrating in the system, you can use a 1fs bond vibration.
what methods can we use to overcome hydrogen bond vibrations in MD?
can restrain the bond vibration to the equilibrium bond legnth
we can convert the hydrogens to deuterium
should vibrate slower but will have the same chemistry
this allows you to use a bigger time step
how do we choose the temperature in MD?
this sets the momentum to zero
this is summed for all particles in the system to get the temperature
not all integration algorithms are reliable estimation of the temperature
we need an integration algorithm to reliably reproduce the temperature

What are periodic boundary conditions in molecular dynamics?
Periodic boundary conditions are used to stop a simulation box behaving like an isolated box with vacuum around the edges. The central box is replicated infinitely in all directions, so when a particle leaves one side of the box, an identical particle from the neighbouring image appears to replace it. This means atoms in the central box always behave as if they are surrounded by more particles, rather than empty space.
Why do we use periodic boundary conditions in MD simulations?
Real biological systems do not exist in a vacuum, so a simulation box with open edges would give unrealistic behaviour. Without periodic boundaries, atoms at the edge of the box would experience fewer interactions than atoms in the middle, which would distort the results. Periodic boundary conditions make the environment more realistic by keeping the central box surrounded on all sides, especially important for things like proteins in water.
Why can periodic boundary condition boxes be different shapes, and why is that useful?
The box does not have to be cubic, as long as the shape tessellates perfectly in space. This means shapes such as octahedra can be used instead of a cube. For a protein in solution, an octahedral box can reduce the amount of unnecessary water in the corners, which lowers the number of atoms in the system and saves computation time. This matters because molecular dynamics is expensive and inefficient, so reducing unnecessary atoms helps simulations run faster without losing much accuracy.
how do we set up a system in MD?
take structure and do energy minimisation
put in the environment you want - eg.; slowly add lipids
for membrane environments, we make cut a hole in the membrane, add the protein
we can then fix the protein position and run a simulation for a small amount of time - this lets lipids move in and pack
add water (and remove any that gets into the membrane)
this uses a sequential slow method
why do we fix protein positions when setting up MD simulations?
if the protein isnt fixed, it may move to fill the gaps - this is not a realistic protein conformational change, it is instead because we cut a hole
what other things may we need to consider/edit in MD?
n the structure itself, there could be crystallographic waters
it wont be solve din a membrane, there may we waters that need removal
there may be detergent molecules that need to be removed
there may be regions missing - eg. flexible loops may not be there in a rystal structure
structure is unlikely to be high enough resolution to determine hydrogens
mutations may have been done to immobilise specific regions
structures may have been solved with ligands
orientation of the protein in the membrane - may need to determine this
issues with x-ray
describe timescale limitations in MD?
MD is inefficient, there are always timescale limitations
Currently, state-of-the-art simulations will not exceed 1-10 microseconds of ‘real time’.
there are some labs that can get longer - eg. chips may be optimised to do MD
If CCs take too long to happen, you may not be able to see with conventional MD
Encounters between two molecules for which diffusion is necessary will also occur on a slower timescale than microseconds
for all atom MD simulations, this will not be possible
we can use algorithmic advancements or coarse grained to overcome this
these can also be combined
eg. can start with low res coarse grained to get the molecules to come together and once they are together, we convert into all atom, put details back in and then look at fine detail
Enhanced sampling methods are needed to overcome (in part) this limitation.
What can we do first to assess simulation quality?
look at the simulation
it can be easy to start to quantify, but a lot of CPU is wasted by running simulations that have blown up
what basic checks can we do to check that a simulated system is behaving as it should
simple distribution of thermodynamic of thermodynamic properties
energy stability
fluctuations of atomic positions, and whether we would expect to see them
→ we can compare these to an infinite distruibtion, eg. we would expect the temperature to be gaussian
what do we have to assume to do basic checks of a simulated system?
we have to assume that the simulation reflects an equilibrium
when is a state in equilibrium?
a state when all of the properties become independent of the simulation time
why can it be difficult to determine equilibrium for a system?
different variables for a system could have different equilibration times, and so may not all be in equilibrium
what is equilibrium for a given property?
we can say that the system is in equilibrium with respect to the variable X when the mean and variance of X are independent of time
What assumption do we rely on when estimating properties from MD? why is this a problem?
We have to rely on the assumption that we sample enough parts of the phase space around the native structure to obtain reliable estimates of our property of interest.
There is no way to detect whether we have missed important parts of the phase space
how can we overcome the inability to know whether we have missed important parts of phase space in MD?
guess or know some of the slow motions likely to be important
do multiple repeats better than one long simulation
do block averaging to reveal problems
looking at how a property changes as a function of simulation time
how can multiple repeat simulations be better than a long one?
eg. do 4x250ns simulations rather than 1µs simulation
this is because when each independent simulations are started, they have different initial starting conditions - they go off on different paths
this means they are less likely to get stuck in the same single metastable state, which might happen for 1 long simulation
what are the two types of error in MD?
systematic errors
statistical errors
what are the types of systematic errors for MD?
rounding up errors in the maths of the algorithms themselves
things missing from the algorithms
eg. may have missed cation pi interactions explicitly in the forcefield
machine precision can be an issue
when you compute using CPU, it can only compute to a defined number of decimal points, and will then round
this rounding up error will accumulate over time
becuase there are so many time steps, this will eventually add up and accumulate
parameters
we have to parameterise the force field interactions
this can be problematic, particularly for transition metals
what are the types of statistical errors for MD?
errors are reported as a standard error of the mean
we average the trajectory vs the true average, from an infinite simulation
what does block averaging do?
block averaging provides good estimate of he SEM of the entire ensemble
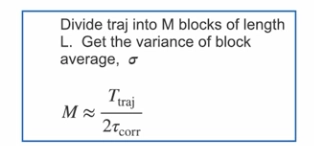
describe how we do block averaging?
we take are simulation and divide it into M blocks of length L
we then calclate the variance of the block average
by combining sigma and M, we get the standard error of the mean for the whole ensemble
we need the blocks of a certain length so there aren't too many correlated frames in the block
we need to make sure the last frame isnt predictable from the first frame
so each block should contain more blocks than the correlation time
equations for block averaging?
in what ways can we determine sample averages for whole trajectories?
stratified systematic sampling
stratified random sampling
coarse graining
how does stratified systematic sampling determine averages for a whole trajectory?
take single value from each block
how does stratified random sampling determine averages for a whole trajectory?
take single value from each block at random
how does coarse graining determine averages for a whole trajectory?
average for each block is determined and then the average of those is taken
Coarse-graining used for thermodynamics properties whereas stratified sampling often used for structural properties (eg. g(r) - the radial distribution function
in which cases can we compare between macroscopic experimental and MD simulatioN>
We can make the comparison as long as the ergodic principle is obeyed
this says that the time average is the same as the ensemble average
we assume that the molecule visits all of the different states seen in the real experiment
when can we assume the ergodic principle is obeyed?
to do this, we do our simulations in a way to mimic the experimental conditions
eg. constant temperature, pressure and a fixed numbr of particles
→ this is an isothermal, isobaric ensemble
what can MD be useful for in terms of structures?
folding small proteins
many proteins are much bigger, so there are a lot of things that could go wrong
this means that folding bigger proteins is much more challenging
so may only become interesting if you are looking at the folding dynamics
what can be observed if you simulate for long enough?
If you simulate long enough, you can watch ligand bind
very rare to do - not done routinely
requires a lot of computation
why do coarse grained simulations give longertime scales?
fewer particles - less things to compute → so gives us longer time scales and larger systems
what are the advanced MD topics? what are they useful for?
potential of mean force/umbrella sampling
Steered MD
Metadynamics - for improved sampling
ΔG of binding - for drug affinity predictions
why may it be difficult to determine properties like drug permeation in MD?
we could run the simulation for a long enough time to try observe this
the chances of being able to osberve this is very small
in the energetic profile, we look at the free energy as a function of distance across the bilayer
we can see that it starts off, and there is a small barrier to gett9ng throguh the headgroups
there is then a large well - very favoured!
there is a small barrier between the wells, so may flucturate between the two
but getting out of the wells will be very unfavoured