Mucopolysaccharidoses and Peroxisomal Disorders - Biochem Genetics
1/43
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
44 Terms
Mucopolysaccharidoses (MPS): Metabolic Pathways
Group of diseases caused by mutation in enzymes that are necessary for the breakdown of
Heparan Sulfate - Abundant in Brain
Keratin Sulfate - Abundant in Skelton
Dermatan Sulfate - Abundant in Skeleton
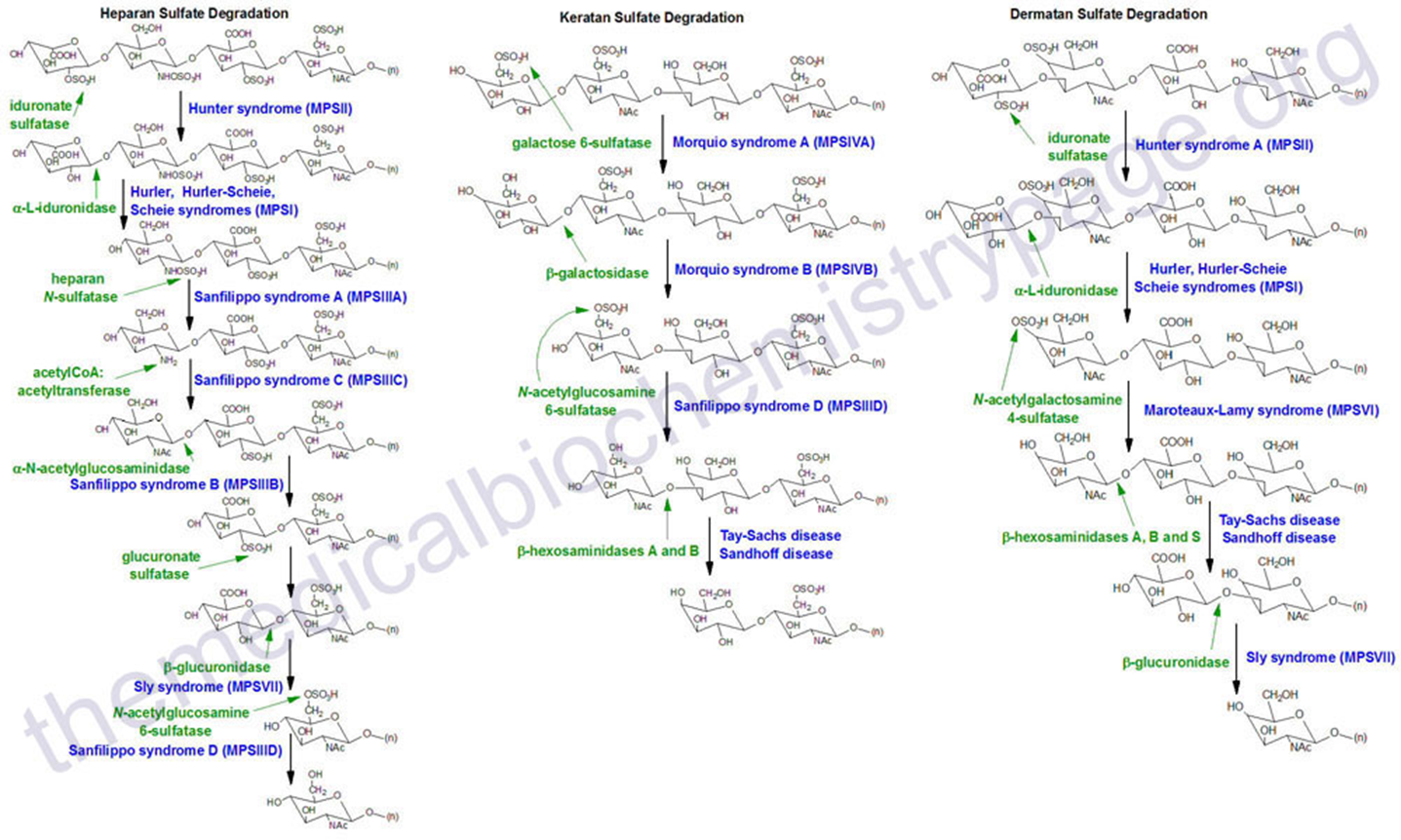
Mucopolysaccharidoses (MPS): Overview
7 Types of MPS
All are autosomal recessive:
Exception MPSII - Hunter syndrome → X-linked Recessive
All are progressive and Asymptomatic at Birth
Exception MPS VII - Sly Syndrome → can present at birth
All Exhibit Abnormally High Urine MPS (GAG) levels
Except GAGs may be only intermittently high in some patients
MPS: Major clinical feaures
“Coarse” Facial Features
Macrocephaly
Flat Nasal Bridge
Large Lips
Broad Mouth
Puffiness around eyes
Coarse “Thatch-Like” Hair
Dysostosis Multiplex: multiple bone deformities
growth retardation
Spinal gibus deformity
“Claw” hand deformity
Mucopolysaccharidoses (MPS): Organ Involvement
Gastroenterologic
Hepatosplenomegaly: enlarged liver and spleen, but no dysfunction
Bowels: Umbilical and inguinal hernias
Cardiopulmonary (collection in muscle)
Cardiomyopathy
Valvular Heart disease
HTN
Coronary Artery disease
Thick Tongue/Airways
Obstructive airway deiase
Sever sleep apnea
Mucopolysaccharidoses (MPS): ENT, Hearing, Vision
Chronic upper respiratory Infections: sometimes first presentation, but nonspecific
Hearing Loss: conductive and sensorineural
Ocular Manifestations
***Corneal clouding***: GAG deposition in Cornea
Retinal degeneration
Glaucoma
Optic Nerve Disease
Mucopolysaccharidoses (MPS) Neurological Sequalae
Neurodegeneration
developmental delays and stagnation
progressive mental deterioration
Communicating Hydrocephalus
contributes to macrocephaly: shunting alleviates symptoms
Peripheral nervous System
Nerve compression: carpal tunnel, Spinal nerve compression
Mucopolysaccharidoses (MPS I): Overview
Deficiency of the a-L-Iduronidase enzyme → IDUA Gene
Spectrum of severity
Hurler: Severe affect → neurological deterioration
Hurler-Scheie: Moderately affect psychically , but no neurological deterioration (normal intelligence)
Scheie: Attenuated affect → neurological deterioration not present
MSP IH (Hurler Disease) classic progression
Diagnosis made at 15 months of age, at which time he had developmental delay, hepatomegaly, and skeletal dysostosis
In the photo, at age 4, the patient had short stature, thick tongue, persistent nasal discharge, stiff joints, claw hand & hydrocephalus
Verbal language skills deteriorated to four or five words. The patient had severe hearing loss and wore hearing aids.

MPS 1 Severe: Outcome without treatment
Features often noted before 1 Year of age → non-specific findings
Umbilical/Inguinal Hernia
Frequent Upper Respiratory Infections
Progressive features often noted from 1yo onward
Coarse facial features and corneal clouding
Dysostosis Multiplex and Organ involvement
Developmental delay, Stagnation, Neurodegeneration
Death often by 10 years of age
MPS 1 Severe: Treatment
Hematopoietic Stem Cell Transplant
If started before 2 years of age → increases survival, but does not present neuro, cardiac, and skeletal features
Recombinant IV Enzyme Replacement Therapy
helps NON-NEUROLOGIC complications → but cannot cross the BLOOD-BRAIN barrier (like most lysosomal storage disorders)
MPS 1 Attenuated: Outcome without treatment
Features usually evident by 3-10 years of age
Milder but slow progressive Dysostosis Multiplex, Facial Coarsening
Cloudy cornea, Hernia, Heart Valve
Normal cognitive outcome to mild learning disability
Normal lifespan to death in 2nd/3rd decades
MPS 1 Attenuated: Treatment
Hematopoietic Stem Cell Transplantation:
rarely done because enzyme replacement is so much more effective
Recombinant Enzyme Replacement Therapy
Aldurazyme reduces non-neurologic complications and is most effective for attenuated MPS I
MPS I: Diagnosis
Urine GAG Excretion
Elevated total GAG levels
Elevated Heparan and Dermatan Sulfate (also seen in MPS II)
Confirmation: Alpha-L-Iduronidase Enzyme Activity
WBCs, Plasma, Fibroblasts, Amnio, CVS
IDUA Gene molecular analysis
NBS: only done in Missouri
MPS II (Hunters): Overview
Iduronate Sulfatase deficiency → mutations in the IDS gene
Dermatan and Heparan Sulfate buildup
ONLY X-Linked
A Severe and Mild form:
Dystosis multiplex
Organomegaly
Mental retardation → death before 15yo
NO CORNEAL CLOUDING
MSP II Severe (Hunter syndrome): Calssic progression
Diagnosis suspected at 1 year of age because of facial appearance, but urine mucopolysaccharide screen was negative at that time.
Enzymatic diagnosis then pursued at age 22 months after a family history of a maternal uncle with mental retardation and coarse facial features became known to the parents.
In the photo, at 6 years of age, the patient shows hepatomegaly, umbilical hernia, joint stiffness, claw-hand, severe hearing loss and severe mental retardation. There was no corneal clouding.

MPS II Severe: Outcome without treatment
Features often noted before 1 year of Age
Umbilical hernia
Frequent upper respiratory infection
Progressive Features often noted form 1 year of Age onward
Coarse facial features WITHOUT CORNEAL CLOUDING
Dysostosis Multiplex and Organ Involvement
Developmental delay, stagnation, neurodegeneration by 6-8 yo
Death often by 10-20 yo
MPS II Severe: Treatment
Hematopoietic Stem Cell Transplantation
better if done before 2 yo, but outcome unclear
Recombinant Enzyme Replacement
Elaprase reduced non-neurologic complications (does not cross blood/brain barrier)
MPS II Mild/Attenuated: Outcome without Treatment
Features usually evident by 10 years of Age
Milder but slowly progressive: Facial coarsening, Dysostosis Multiplex
Organomegaly, Hernia, Heart valve disease
Normal cognitive outcome / minimal learning disability
Range from Normal lifespan to death in 2nd/3rd decades
MPS II Mild/Attenuated: Treatment
Hematopoietic Stem Cell Transplantation: rarely used b/c enzyme replacement better
Recombinant Enzyme replacement therapy:
reduces non-neurologic complications → most effective for Mild MPS II
MPS II Severe: Diagnosis
Urine GAG Excretion
Elevated total GAG levels
Elevated Heparan and Dermatan Sulfate (like MPS I)
Iduronate Sulfatase Enzyme Activity (not useful for carrier screening)
WBC, Plasma, Amnio, CVS
IDS Gene molecular Analysis
MPS III (SanFallipo Syndrome): Overview
4 types → 4 Enzymes → 4 Genes
All involved exclusively in Heparan Sulfate
Main clinical Manifestations are NEUROLOGIC
NO COARSENING OF FACIAL FEATURES
NO EFFECTIVE TREATMENT (enzyme replacement cannot cross the blood brain barrier)
Most Common MPS, but VASTLY underdiagnosed
MPS IIIA: Classic Presentation
The patient had normal developmental skills until about 5 years of age, at which time she was toilet trained, was able to feed and dress herself, and spoke in complete sentences. She was learning to read and write.
In the photo, at age 7, she had a relatively normal physical appearance with mild hepatomegaly, but now had significant loss of cognitive function.
At age 11, she was able to feed herself but had lost all verbal skills.

MPS III Sanfilippo: Typical Natural History
Mainly REGRESSIVE
Early childhood behavior changes (ADHD, aggression)
Cognitive plateau → Progressive Neurodegeneration
****Little to no Organomegaly, Corneal Clouding ****
Death often before 20 yo (Cardiopulmonary Effects)
MPS III Sanfilippo Type A-D: Diagnosis
Suspicious if there is REGRESSIVE AUTISM
Elevated Urine GAGs → HEPARAN SULFATE ONLY
Need Enzyme/Molecular Analysis to confirm and distinguish Types !-D
MPS IVA (Morquio)
2 types of enzymes → 2 types of genes → 2 forms of disease
A: more common → Kertan sulfate + Chondroitin 6-Sulfate
B: Kertan sulfate only
Normal Intelligence → No NEUROLGIC EFFECTS
But very specific Dystosisis and typical Cloudy Cornea
MPS IVA: Skeletal Findings
Ulnar Deviations of the Wrists
Shortened forearms
Pectus Carinatum
Genu Valgum (Knock-Knees)
MPS IVA (Morquio): Outcome without treatment
Severe form: often leads to death by 30-40yo
Features often noted between 1-3 yo: skeletal findings
Progressive features
Skeletal short stature, skeletal deformation, spinal stenosis,
Non-Skeletal
Cornel Clouding, Valvular heart Disease, Mil Hepatomegaly, Hearing impairment, Sleep Apnea, Severe Respiratory Insufficiency
Mild-Form": Mlder, later onset
***NO INTELLATUAL DIABLITY WITH ANY FORM OF DIEASE***
MPS IVA (Morquio): Treatment
Recombinant Enzyme Replacement Therapy: biological and functional improvements → particularly helpful for this condition
Surgical Procedures
MPS IVA (Morquio): Diagnosis
Urine GAG Excretion
Elevated Total GAG levels
Elevated Keratan and Chondroitin 6-Sulfate -
ONLY Keratan Sulfate elevate in MPS IVB (B-Galactosidase Gene)
N-Acetylgalactosamine 6-Sulfatase Enzyme Activity
WBCs, Fibroblasts, Amnio, CVS
B-Galactosidase measured as well
GALNS Gene molecular analysis
MPS VI (Maroteaux-Lamy): Overview
Arylsulfatase B deficiency → mutations in ARSB gene
Dermatan Sulfate builds up
Coarsening of features, Skeletal Dysostosis
Progressive skeletal weakness
BUT NO NEUROLGIC AFFECTS → can live normal length lives
****LEAST COMMON MPS***
MPS IV (Marateaux-Lamy): Typical natural history
Decelerated growth after 1st year → short stature/dwarfism
Progressive
facial coarsening,
corneal clouding
Cardio-Respiratory disease
Organomegaly
Dysostosis Multiplex
NORMAL INTELLGIENCE
Death in Teens → SEVERE CASES, longer surival in midl cases
MPS IV (Marateaux-Lamy): Diagnsois
Elevated GAG → Dermatan Sulfatae
Enzyme/Molecular Analysis for confirmation
MPS IV (Marateaux-Lamy): Treatment
IV Recombinant Enzyme Replacement Therapy
Biochemical and phenotypic improvement —> VERY EFFECTIVE
MPS VII (Sly Syndrome): Overview
Rarest MPS:
Often starts In-Utero leading to Fetal Demise
Evident at Birth Except in Mildest Cases
Etiology: B-Glucuronidase (GUSB) gene mutation
Dermatan, Heparan and Chondroitin 6-Sulfate Accumulate
Features
Dysostosis Multiplex, Organomegaly, Coarse Features, Neurodegeneration, Corneal Clouding
Death by 20s even in mild cases
Treatment
Supportive care and Clinical Trials (ERT in Phase III)
Peroxisome Disorders
Ubiquitous single membrane organelles catalyzing a variety of reactions
Two Types
Peroxisome biogenesis disorders: faulty construction of peroxisomes → all reaction are faulty (PEX Disorders)
Sing gene enzyme disorder → specific reactions are affected within the peroxisome (only one important: X-linked adrenoleukodystrophy)
X-Linked Adreno Leukodystrophy
Mutations in the ABCD1 Gene → Produces ATP transport protein that transport the very long chain fatty acids into the peroxisomes so they can be degraded via beta oxidation
Found only on x chromosome → only boys affect: X-Linked Recessive
Accumulation of very long fatty chains in the body: especially the nervous system + the adrenal gland
3 Major phenotypes
Childhood leukodystrophy
Adreno myeloneuropathy
Addison’s Disease
Untreated X-ALD: Childhood Cereberal Leukodystrophy
The wrost of the 3 pehnotypes
Males with intial normal devleoment for 4-10 years
Frist symptoms
Behaviour and attention
School difficualt, handrwrting issues
Visual loss, comerphension difful
Follwed by rapid neurlogic derion
White matter abnormalties
adrenocorticol insufficney
Total disability and death within 2 Year
Untreated X-ALD: Adrenomyeloneuropayh
Males iwth intially nromal health for 20-40 years
Then develop
leg weakenss, bowle/bladder, and sexual dysfunction
Followed by adrenocotical insufficney in 70% and progressive cogntive and behavioural impairment in 20% (Can be lethal)
BRONZING OF THE SKIN
Untreated X-ALD: Isoalted Addisons
10 % of patients
Primarily Adrenocortical Insufficiency
High ACTH → Bronze Skin, Low corticosteroids and Mineralocorticoids → Weakness, Vomiting, Coma
Wide Range of Age at Onset (2yo-Adult)
Late-Onset Mild Myeloneuropathy may also develop
X-Ald: Diagnosis
Diagnostic Tests: Plasma Very Long Chain Fatty Acid (VLCFA) Levels
Abnormal in 99% of affected males, but not as well for affect females or femal carriers
Genetic Testing: ABCD1 gene mutation
Prenatal Tests: Molecular, VLCFA levels on Amnio/CVS
Other routine test
Brain MRI
Adrenal function testing