Farmacologia I - Brenner Cap 2 Farmacocinética: lo que el organismo le hace al fármaco
1/72
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
73 Terms
Farmacocinetica
Efecto del organismo al fármaco
El estudio de la distribución del fármaco en el organismo y se centra en los cambios observdos en la concentración plasmática del farmaco
Para cualquier fármaco y dosis determinados, la concentración plasmática del fármaco aumentará y descenderá en función de la velocidad de cuatro procesos
*Liberación
Absorción
Distribución
Metabolismo
Excreción
Absorción
Es el desplazamiento del fármaco desde el lugar donde se administra hasta el torrente circulatorio
Requiere que los fármacos atraviesen una o varias capas de células y de membranas celulares
Distribución
Es el el proceso por el cual el fármaco abandona el torrente sanguíneo y se reparte por los órganos y tejidos del cuerpo
Metabolismo o biotransformación
Es el proceso de conversión de un fármaco en uno o varios metabolitos, principalmente en el hígado
Excresión
Es la expulsión del fármaco o sus metabolitos del cuerpo, principalmente por los riñones y la orina
Eliminación
Conjunto de procesos del metabolismo y excresión.
El proceso de absorción de fármacos forma parte de todas las vías de administración excepto de la vía/s: Elige todas las que apliquen
a. Renal
b. Intramuscular
c. Tópica
d. Intravenosa
c. Tópica: El farmaco se aplica directamente en el tejido diana
d. Intravenosa: El farmaco se introduce directamente en el torrente sanguineo
Los fármacos que se administran via _______ y ________ evitan la barrera epitelial, por lo que se absorben más facilmente a través de los espacios existentes entre las células endoteliales de los capilares.
Intramuscular y Subcutáneo
En el intestino, pulmones y piel, los fármacos deben atravesar una:
a. Capa de células epiteliales con uniones estrechas
b. Capa de células epiteliales con membranas basales fenestradas
c. Capa de células epiteliales con GAP junctions
a. Capa de células epiteliales con uniones estrechas
V o F: Los fármacos se enfrentan a una barrera más importante en la absorción tras la administración oral que tras la administración parenteral.
V
La mayoría de los fármacos se absorben por:
a. Difusion facilitada
b. Difusión pasiva
c. Transporte activo primario
d. Transporte activo secundario
b. Difusión pasiva - A traves de una membrana para llegar a la circulación
V o F: La velocidad de absorción es proporcional al gradiente de concentración del fármaco a través de la barrera y a la superficie disponible para la absorción en ese lugar
V
Ley de Fick
La velocidad de difusión de un fármaco a través de una membrana es directamente proporcional al:
área de superficie,
gradiente de concentración,
coeficiente de difusión,
e inversamente proporcional al grosor de la membrana (y tamaño de la molécula)
¿Qué factores aumentan la difusión de un fármaco según la Ley de Fick?
Mayor área superficial
Mayor gradiente de concentración
Mayor liposolubilidad
Menor grosor de membrana
Ley de Fick y Gradiente de concentración
Mientras mayor sea la diferencia de concentración:
→ más rápida difusión.
Ejemplo
Después de administrar una dosis oral:
concentración alta en intestino
concentración baja en sangre
El fármaco difunde hacia sangre.
¿Qué es el coeficiente de difusión?
Es la medida de qué tan fácilmente una sustancia atraviesa una membrana o se desplaza en un medio
Depende de
Tamaño moleculas: Moléculas pequeñas → difunden más fácilmente.
Liposolubilidad: Mientras más liposoluble sea el fármaco → mayor coeficiente de difusión.
Otros: Temp → Favorece; Viscosidad → Dificulta
Un fármaco con alto coeficiente de difusión:
atraviesa membranas rápido
se absorbe más fácilmente
llega más rápido a tejidos
¿Qué características aumentan el coeficiente de difusión de un fármaco?
Tamaño pequeño
Alta liposolubilidad
Forma no ionizada
Mayor temperatura
Los fármacos pueden ser absorbidos de forma pasiva atravesando las células mediante:
Difusión lipidica
Difusión acuosa
La difusión lipídica
Es un proceso mediante el cual el fármaco se disuelve en los componentes lipídicos de las membranas celulares.
Este proceso se ve facilitado si el fármaco es muy liposoluble duhh
La difusión acuosa
Se produce por el paso a través de poros acuosos presentes en las membranas celulares.
*Dado que la difusión acuosa se limita a los fármacos con un bajo peso molecular, hay muchos medicamentos que son demasiado grandes para poder absorberse por este proceso.
El transporte activo
Precisa una molécula transportadora y consume energía, proporcionada por la hidrólisis del enlace fosfato terminal de alta energía de la adenosina trifosfato (ATP).
El transporte activo permite transferir fármacos en contra del gradiente de concentración.
La difusión facilitada
Necesita una molécula transportadora, pero no consume energía.
Por este motivo, los fármacos o sustancias no pueden transferirse en contra del gradiente de concentración, pero difunden más rápido que en ausencia de la molécula transportadora
V o F: Muchos fármacos son ácidos o bases fuertes que existen en forma tanto ionizada como no ionizada en el organismo
FALSOOO, son acidos o bases debiles
V o F: Solo la forma no ionizada de estos fármacos es lo bastante soluble en membranas lipídicas como para atravesar las membranas celulares.
V
La proporción entre las dos formas (ionizadas y no ionizadas )en un lugar específico influye en
Velocidad de absorción
Distribución
Eliminación
Los ácidos débiles (HA) vs Las bases débiles (B)
Los ácidos débiles (HA) ceden un protón (H+ − ) → Forman aniones
Las bases débiles (B) aceptan un protón para formar cationes (HB+)
Con ácidos débiles, la forma protonada es la:
a. No ionizada
b. Ionizada
a. No ionizada, por eso puede difundir
Con bases débiles, la forma protonada es la:
a. No ionizada
b. Ionizada
b. Ionizada, por eso NO puede difundir
La pK a de un ácido débil o de una base débil es
El pH en el que existe la misma cantidad de la forma protonada que de la forma no protonada. Se puede utilizar la ecuación de Henderson-Hasselbalch para determinar la proporción entre las dos formas:
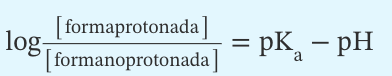
V o F: La forma no protonada de un ácido débil se encuentra no ionizada, mientras que la forma protonada de una base débil se encuentra no ionizada
Falso
La forma protonada de un ácido débil se encuentra no ionizada
La forma protonada de una base débil se encuentra ionizada
Match
pH = pka
pH < pKa
pH > pKa
a. Existe una cantidad idéntica de la forma protonada y de la no protonada.
b. Predomina la forma protonada
c. Predomina la forma no protonada.
pH = pka: a. Existe una cantidad idéntica de la forma protonada y de la no protonada.
pH < pKa: b. Predomina la forma protonada
pH > pKa: c. Predomina la forma no protonada.
Por qué los ácidos débiles se absorben más rápidamente en el estómago que las bases débiles?
En el estómago, donde el pH es de 1, los ácidos y las bases débiles se encuentran fuertemente protonados. En este lugar predominan la forma no ionizada de los ácidos débiles (pK a = 3-5) y la forma ionizada de las bases débiles (pK a = 8-10).
Por qué las bases débiles se absorben más rápidamente en el intestino que los ácidos débiles?
En el intestino, donde el pH es de 7, la mayoría de las bases débiles también se encuentran ionizadas, pero mucho menos que en el estómago, por lo que las bases débiles se absorben más rápidamente en el intestino que en el estómago.
Los ácidos débiles se absorben mejor en medios:
Ácidos, porque predomina la forma no ionizada de estos
¿Los fármacos se distribuyen uniformemente en el cuerpo?
No. Algunos permanecen principalmente:
en plasma,
en líquido extracelular,
o se acumulan en tejidos específicos.
¿Qué es el atrapamiento iónico?
Es la acumulación de un fármaco ionizado en un compartimiento debido a diferencias de pH.
¿Qué característica permite que un fármaco entre fácilmente a las células?
La liposolubilidad, porque facilita atravesar membranas celulares.
¿Qué son los transportadores ABC (ATP Binding Cassette)?
Son proteínas dependientes de ATP que expulsan fármacos fuera de las células → Se oponen a la distribución de lor fármacos por los tejidos
¿Qué es la glucoproteína P (P-gp) y dónde se encuentra?
La Pgp se exLa glucoproteína P (P-gp) es una proteína transportadora de membrana perteneciente a la familia de los transportadores ABC (ATP Binding Cassette), que utiliza ATP para expulsar fármacos fuera de las células y disminuir su concentración intracelular.
Se localiza principalmente en:
intestino,
barrera hematoencefálica,
hígado,
riñón,
placenta.
Funciones de la Pgp e importancia para la farmacología
Funciones e importancia:
disminuye absorción intestinal de medicamentos,
limita el paso de fármacos al cerebro,
favorece eliminación biliar y renal,
protege tejidos frente a tóxicos,
participa en la barrera hematoencefálica y placentaria,
causa resistencia a quimioterapia al expulsar antineoplásicos de células tumorales.
La inhibición de P-gp por fármacos como: amiodarona, eritromicina, verapamilo, propranolol,etc, aumenta la concentración tisular y plasmática de medicamentos, elevando sus efectos y riesgo de toxicidad.
Lista los facores que afectan a la distribución
Vascularización del órgano
Unión a proteínas plasmáticas
Tamaño molecular
Liposolubilidad
¿Cómo afecta la vascularización de un órgano a la distribución de los fármacos?
La distribución depende de la cantidad de flujo sanguíneo que recibe cada órgano.
Los tejidos altamente perfundidos:
cerebro,
corazón,
hígado,
riñón,
reciben los fármacos rápidamente, produciendo un inicio rápido del efecto. Los tejidos menos perfundidos:
músculo,
piel,
hueso,
grasa,
reciben el fármaco más lentamente.
¿Cómo afecta la unión a proteínas plasmáticas la distribución de los fármacos?
Los fármacos se unen reversiblemente a proteínas plasmáticas, principalmente:
albúmina,
glucoproteínas,
β-globulinas.
Solo la fracción libre (no unida) puede:
atravesar membranas,
distribuirse a tejidos,
ejercer efecto farmacológico.
Los fármacos ácidos suelen unirse a albúmina y los básicos a glucoproteínas y β-globulinas. La unión es saturable y algunos medicamentos pueden desplazar a otros, aumentando toxicidad.
¿Cómo influye el tamaño molecular en la distribución de un fármaco?
Las moléculas grandes se distribuyen menos porque atraviesan con dificultad las membranas capilares. Por eso algunos medicamentos, como la heparina, permanecen principalmente en el compartimento plasmático.
¿Cómo afecta la liposolubilidad la distribución de los fármacos?
La liposolubilidad es uno de los factores más importantes para la distribución tisular. Los fármacos liposolubles atraviesan fácilmente membranas celulares y la barrera hematoencefálica, mientras que las moléculas polares e ionizadas penetran con dificultad al SNC debido a las uniones estrechas de la barrera hematoencefálica.
¿Cómo afecta la motilidad gastrointestinal la absorción de los fármacos?
La motilidad gastrointestinal modifica el tiempo de contacto del fármaco con la superficie de absorción. Un tránsito rápido disminuye la absorción, mientras que un tránsito lento puede aumentarla.
¿Cómo influye el vaciamiento gástrico en la absorción?
Mientras más rápido el estómago vacía su contenido hacia el intestino delgado, más rápida suele ser la absorción, porque el intestino tiene gran superficie de absorción.
¿Cómo afecta el pH la absorción de un fármaco?
El pH modifica el grado de ionización del fármaco. Las formas no ionizadas atraviesan mejor las membranas y se absorben más fácilmente.
¿Cómo afecta el flujo sanguíneo esplácnico la absorción?
Un mayor flujo sanguíneo intestinal mantiene el gradiente de concentración y favorece la absorción de los medicamentos.
¿Cómo influyen la formulación y el tamaño de partícula en la absorción?
Las partículas pequeñas se disuelven más rápido y aumentan la absorción. Las formulaciones farmacéuticas, como cubiertas entéricas o liberación sostenida, también modifican la velocidad de absorción.
¿Qué factores fisicoquímicos afectan la absorción de los fármacos?
Liposolubilidad
Coeficiente de partición
Solubilidad
Naturaleza química
Peso molecular
Estos determinan qué tan fácilmente el fármaco atraviesa membranas.
¿Qué es el efecto de primer paso y cómo afecta la absorción?
Es el metabolismo del fármaco en intestino e hígado antes de llegar a circulación sistémica. Disminuye la biodisponibilidad oral.
¿Cómo afectan los alimentos la absorción de medicamentos?
Los alimentos pueden:
retrasar vaciamiento gástrico,
alterar el pH,
unirse al fármaco,
disminuir o aumentar absorción.
Farmacodependencia
La farmacodependencia es un estado en el que una persona desarrolla:
necesidad compulsiva de consumir una sustancia,
pérdida parcial del control sobre su uso,
dependencia física y/o psicológica.
También puede llamarse:
drogodependencia,
dependencia a sustancias.
¿Qué es la dependencia psicológica?
Es la necesidad emocional o mental de consumir una sustancia para obtener placer o evitar malestar.
¿Qué es la dependencia física?
Es la adaptación del organismo a una droga, produciendo síntomas de abstinencia cuando se suspende.
¿Qué es la tolerancia farmacológica?
Es la disminución del efecto de una droga tras su uso repetido, requiriendo dosis mayores para lograr el mismo efecto.
¿Qué es la tolerancia farmacodinámica?
Es la disminución de respuesta debido a cambios en receptores o mecanismos celulares.
¿Qué es la tolerancia farmacocinética?
Es el aumento del metabolismo o eliminación del fármaco, disminuyendo su concentración.
¿Qué es el síndrome de abstinencia?
Es el conjunto de síntomas físicos y psicológicos que aparecen al suspender una droga en una persona dependiente.
¿Qué neurotransmisor está fuertemente relacionado con adicción y recompensa?
Dopamina
Menciona sustancias con alto potencial de farmacodependencia.
Opioides
Alcohol
Nicotina
Cocaína
Benzodiacepinas
Anfetaminas
¿La tolerancia y la dependencia son lo mismo?
¿La tolerancia y la dependencia son lo mismo?
Biodisponibilidad
Es la fracción (F) de la dosis administrada de un fármaco que alcanza la circulación sistémica en su forma activa

-Indica cuánto medicamento realmente está disponible para producir efecto.
Factores que disminuyen biodisponibilidad
absorción incompleta
metabolismo hepático
degradación gástrica
efecto de primer paso
interacción con alimentos
vómitos o diarrea
Factores que aumentan biodisponibilidad
buena absorción intestinal
alta liposolubilidad
poco metabolismo hepático
formulaciones adecuadas
Biodisponibilidad - Relación con vías de administración
IV
100%
Oral
variable
Sublingual
alta
evita primer paso hepático
El volumen de distribución (V d )
Se define como el volumen de líquido en el que debería disolverse una dosis de un fármaco para alcanzar la misma concentración que presenta en el plasma
Volumen de distribución (Vd)
Describe qué tanto un fármaco se distribuye fuera del plasma hacia los tejidos.
-Es un “volumen aparente” que irve para estimar cuánto medicamento permanece en plasma, o cuánto se distribuye hacia tejidos.

Vd pequeño significa:
alta concentración plasmática
poca distribución tisular
El medicamento permanece principalmente:
en sangre o plasma.
Factores que aumentan el Vd
alta liposolubilidad
bajo peso molecular
baja unión a proteínas plasmáticas
alta unión a tejidos
Factores que disminuyen el Vd
gran tamaño molecular
alta unión a albúmina
hidrosolubilidad
confinamiento vascular