genet 270 pt 3
1/12
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
13 Terms
Suppression and reversion overveiw
- Reversion: any mutational process that restores wt to cells that are already carrying a phenotype-altering forward mutation
1) true reversion: a reversion from original to the mutant & back to same wt sequence
2) pseudo-reversion/suppression: we have already seen examples of suppression with the discovery of the genetic code. Pseudo-reversion is a phenotypic but not genotypic revertant
- One important observation by the early work of Streisinger was that some mutagens (proflavine) would revert but only due to a second mutation at a second site
- These are called pseudorevertants. How could they test that?
Streisinger worked on a different gene of T4 phage = lysozyme
Lysozyme is a product of a T4 late gene
It digests peptidoglycan (cleaves glycosidic bonds) to help phage infection. Promotes lysis of bacterial cells
- Lysozyme released from the lysed bacterial hosts can diffuse and affect cells nearby the plaque, however, it is usually in too low a dose to lyse the cells
Except – Streisinger knew that if the plate was exposed to chloroform vapours, the chloroform weakens the cells so the combination of low diffuse lysozyme and chloroform would lyse the cells
No chloroform → wt T4 phage plaques. With chloroform → produces halo around plaques
Thus easy phenotypic screen for phage defective in lysozyme = plaques look wt even w/ chloroform
- What he did?
isolated a set of proflavin(induces indels) induced mutants
the lysozyme mutants were crossed w/ each other to produce intragenic recombinants (some restored wt phenotype = intragenic suppressors)
could also cross out mutations to seperate independant mutant loci thus not true revertant
- Ex One of the crosses (eJ42 x eJ44) resulted in half the recombinant phage producing 50% of the normal wildtype level of lysozyme
thus pseduo-et phage must be double mutants for eJ42 & eJ44
- After growing up huge batches of the double mutant phage, he isolated the protein and tried to sequence it
Could NOT sequence it
Digested it with proteases and separated individual peptides on an ion-exchange column
found = one peptide was always diff in its chemical properties
double mutant showed differences compared to both wt & single mutant
He could sequence the single peptide and the sequence differed between wildtype and the eJ42eJ44 double mutant (5 aa were diff compared to wt but → caused by 2 mutations)
- Conclusion: change was exactly what Brenner & Crick had predicted about how genetic code works
Intragenic suppression of frameshift mutations
- often due to: compensatory indel nearby
- 2 requirements:
2nd mutation must be near original mutation; as length of frameshift sequence increases
Inc total # of amino acid errors
Before nonsense codon appears
Region of protein contains amino acid capable of withstanding change
lysozyme is VERY mutationally tolerant
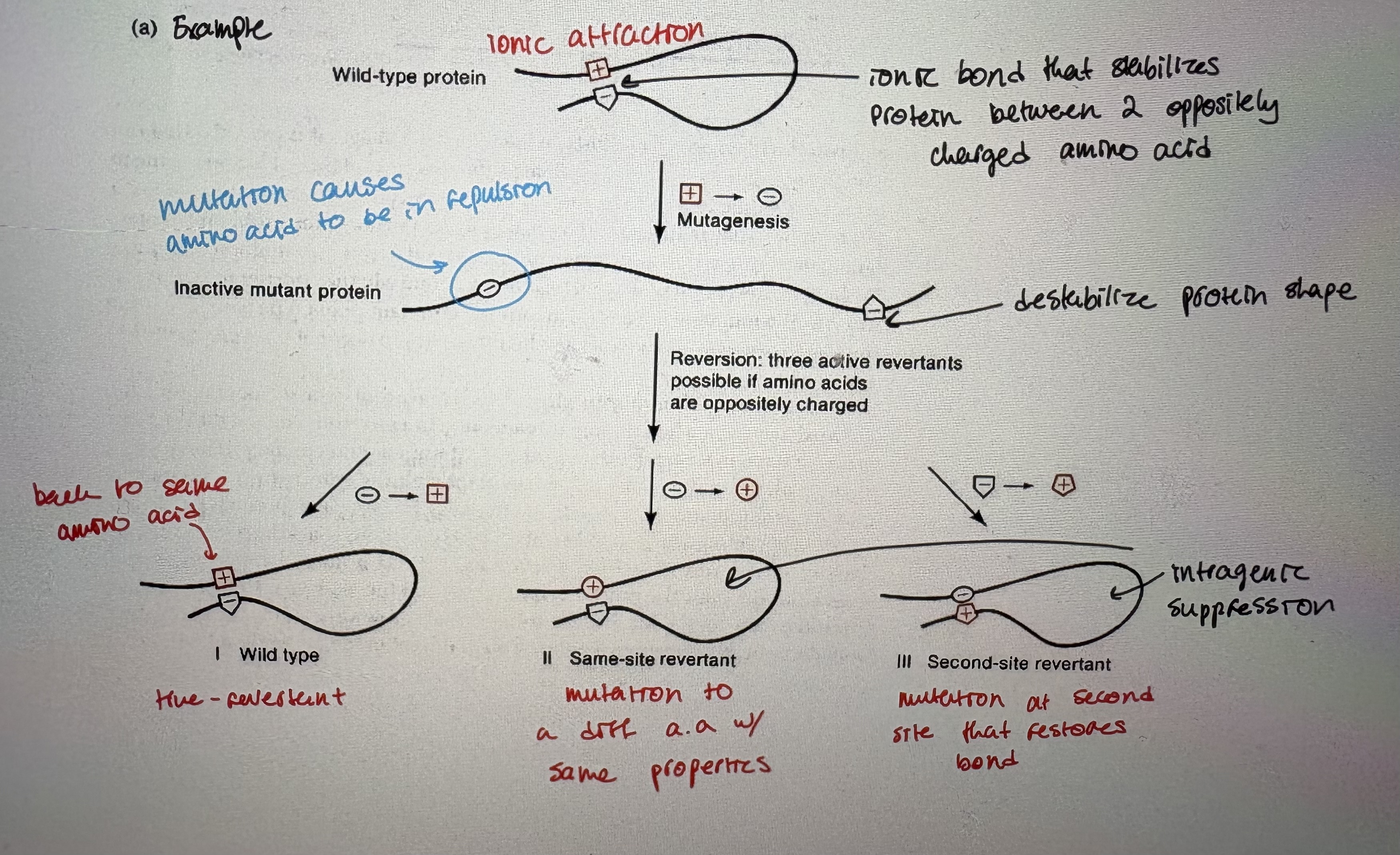
- reversion that occurs as a result of changes within the mutated gene, 2 types: Nonsense & Missense (also other types of intragenic suppression unrelated to frameshift)
Ex (see pic; read)
- missense codons that obliterate protein activity are suppressed by mutations that allow the missense codon to be read as some other amino acid
3 mechanisms: Class l = true reversion. Class ll = mutant tRNA (like nonsense suppressors but tRNAaa1 now encodes tRNAaa2). Class lll = 2nd site revertant
very inefficent (1%) = rare thus phage is the best model
Detection of a nonsense suppressor
- First discovered by Garen’s work on E. coli alkaline phosphatase
It’s the enzyme encoded by the phoA gene in E. coli
Protein is very stable and easy to assay and purify
- A colorless chemical – pNPP (p-nitrophenyl phosphate) is converted to a yellow produce (nitrophenol) by AP
phoA- mutants can be detected by a colorimetric assay = mutants do NOT produce colour when grown on a plate w/ pNPP
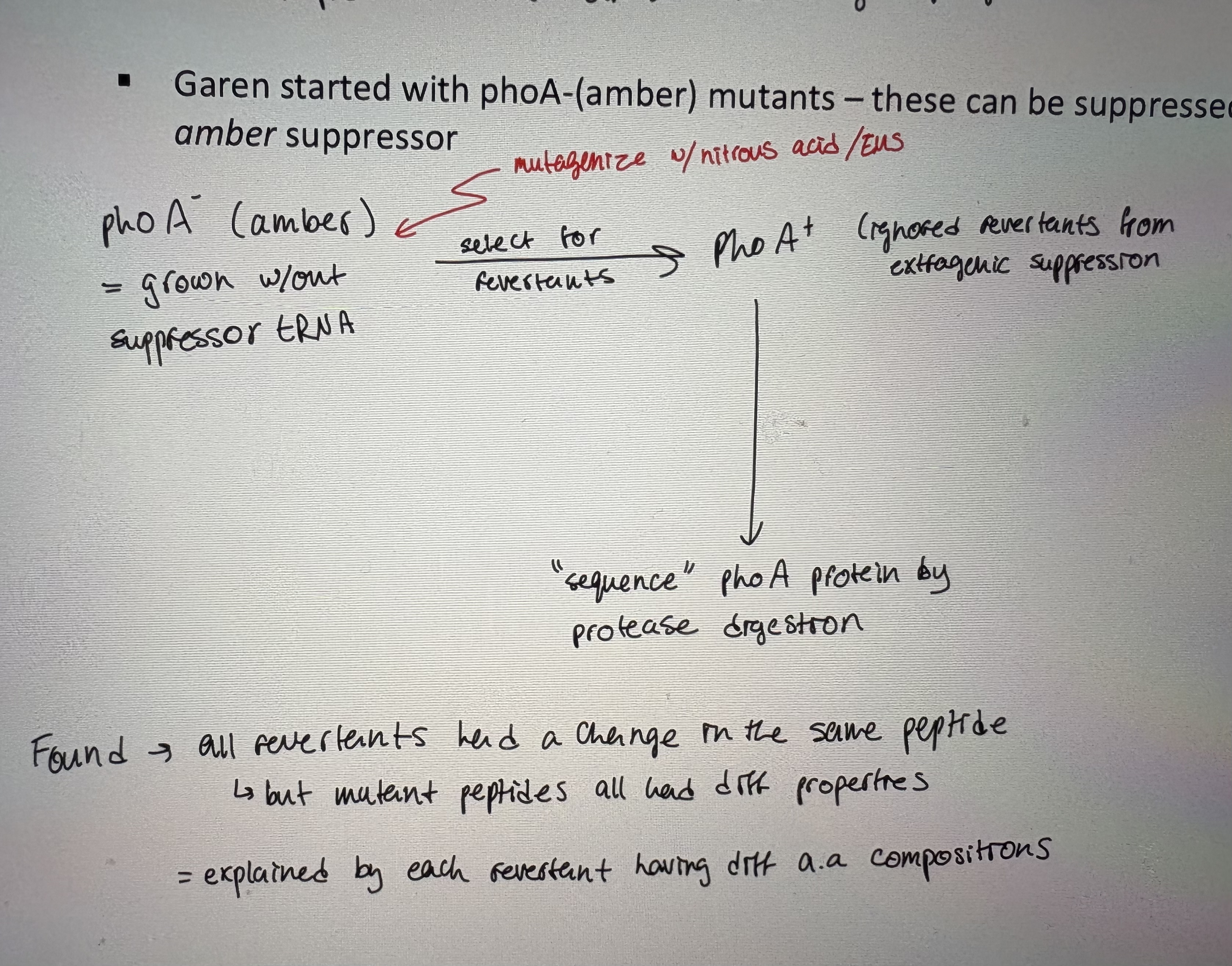
- Garen started with phoA-(amber) mutants – these can be suppressed by E. coli carrying an amber suppressor (see pic)
Found → all revertants had a change in the same peptide, but mutant peptides all had diff properties (explained by each revertant having diff aa compositions)
- The wildtype peptide has several tryptophan amino acids
Most suppressors or revertants (phoA+) had trp replaced by different amino acids
Trp is encoded by UGG codon (non-redundant)
- all phoA- mutant revertants all seemed to involve trp codons
revertants were result of missense mutations to trp codon ex trp → leu, ser, glu, etc (only one base changed)
- Original mutation was due to a single base change
Assumed because it could be suppressed by an amber suppressor
- The revertant was also due to a single base change
Follows that: original codon of intial mutant (UGG-UGA) was one bp diff from original aa at that position
- When looking at what they knew at the time about the genetic code – the only possible changes from UGG that didn’t have an assigned amino acid
identified UGA stop codon
found → UGA can be reverted to missense mutations
- Now we know: tRNA can suppressor original mutation and others → see notes
Nonsense suppression/Intragenic suppression
- nonsense (chain termination) mutations common: lead to production of truncated proteins (usually nonfunctional)
- nonsense suppressors: mutant tRNA that recognizes a stop as a codon
- suppressor strains are not sick even if they ignore 1/3 of stop codons
- due to: mutation in a second gene that can suppressor original mutant
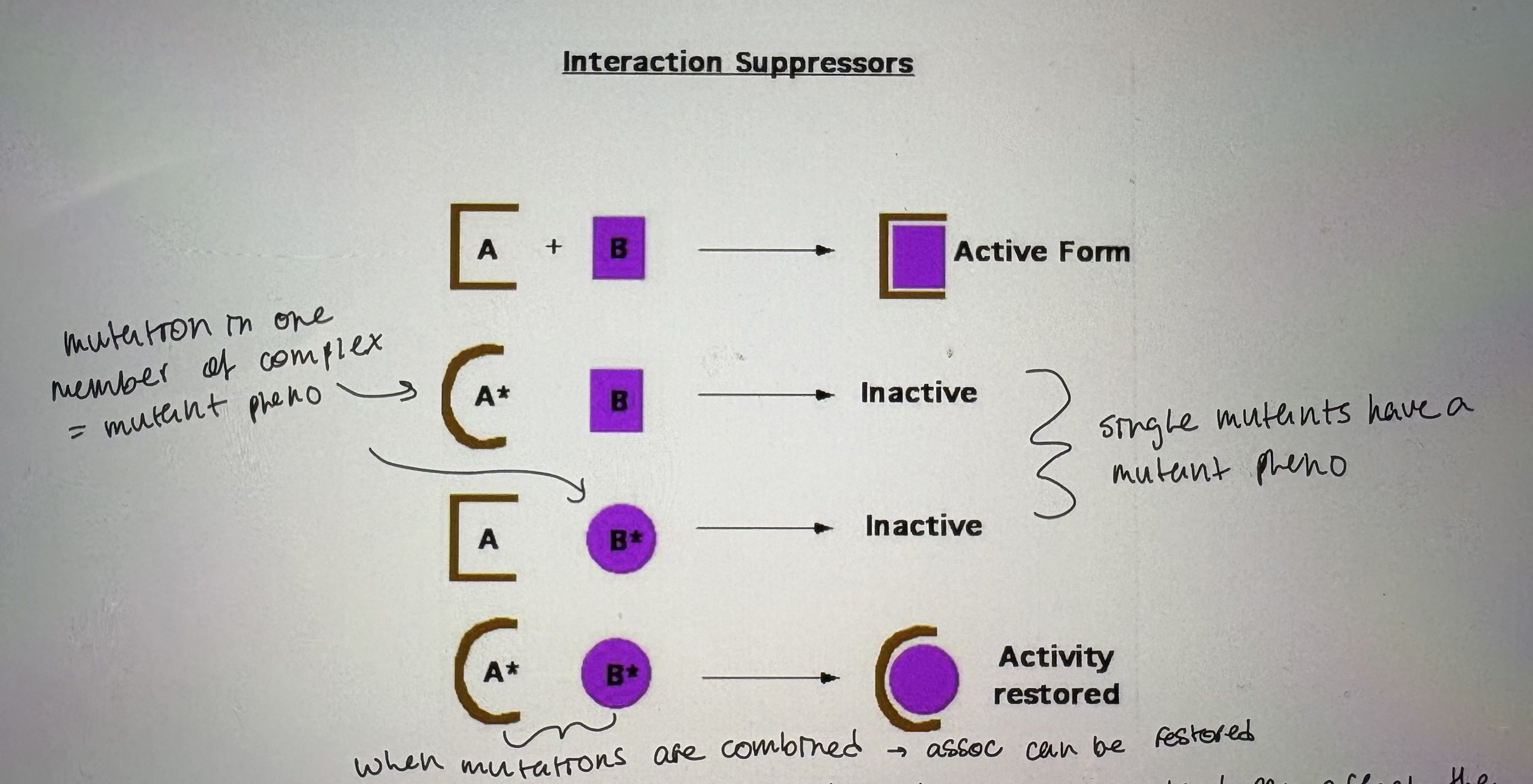
1. Interaction suppressors
- if a protein is part of a larger complex then a mutation in one can disrupt the association (see pic)
mutation in one member of complex = mutant phenotype
single mutants have a mutant phenotype
when mutations are combined → association can be restored
- Often allele specific; diff alleles of original mutant can affect the protein diff
require compatible 2nd mutation to restore interaction
2. Overproduction suppresors
- overcome mutant effect by: changing proportions of original mutant production
- only if: overproduction has a pheno = too much gene production can be deleterious
- often in: suppression mutations are often in regularory domains/factors
- can also affect: induced function mutations
eg. promoter up mutations as suppressors of lacZ- missense mutations
3. Bypass suppressors
- involve 2 mutated genes that belong in diff pathways but are functionally related
- not allele-specific. Ex if mutation causes lose of function in enzyme 2 in one pathway, a compensatory mutation in enzyme 4 in another pathway can replace enzyme 2
4. Physiological suppressors
- general changes in cell physiology that alter general cell physiology to revert to a near normal state
- eg. increasing concentration protein folding factors may suppress many missense mutations that produce proteins that are functional, but unstable
5. Informational suppressors
- cell’s translational machinery changes so that certain mutations can be silenced by changes in gene expression machinery
- cell reads information in mRNA differently
- most often due to tRNA suppressors (not just nonsense suppressor tRNA but missense too)
Uses of reversion frequency/ Ames test
1. To identify the original type of mutation
- different types of mutation revert at different rates
- Single point → Reversion? Possible → very frequent
- Double point → Reversion? Possible → frequent
- Triple point → Reversion? Possible → rare
- Frameshift → Reversion? Possible → frequent
- Deletion → Reversion? Not possible → none
2. To determine cause & effect
- use to determine if phenotypic changes in a mutant due to one mutation or multiple mutations
- to determine if a phenotype is due to a single mutation → look at frequency of reversion
Grow slowly = 2 mutations. Grow normal rate = 1 mutation.
3. To detect mutagens/carcinogens
- use to screen hazardous agents for carcinogenic activity
mutagens are carcinogens, but not all carcinogens are mutagens
Remeber: its dose that makes the poison
- compare reversion freq. to that in absence of compound
- Ames was familiar with the work of Benzer, Crick and Brenner (and others) and knew you could tell a lot about a mutation dependent on its reversion frequency
- Used S. typhimurium cells that had a weak cell membrane (more permeable to chemicals) and defective DNA repair
Exposed to potential and known mutagens
Selected for his+ revertants to test mutational frequency of known compounds
used liver extract = some substances are non-toxic until metabolized at liver
counted revertants
- used on thousands of substances: food additives, pesticides, hair dyes and cosmetics
- reduces animal testing (almost all known carcinogens cause tumor formation in animals)
- Developed a dose-response curve typical for determining whether a chemical is mutagenic
- His results? 90% of peroxide-based hair dyes were mutagenic
- Some potential mutagens that were thought to work like acridines didn’t yield frameshifts yet highly mutagenic
In vitro experiments suggested that these form chemical adducts (or covalent bonds) with DNA
adducts = distort helix & permanent alterations to DNA structure
How the cell deals with these adducts? And other damage
DNA replication overview/ Discovery of DNA pol l
- polymerization of DNA nucleotides → long chains using a single strand of parental DNA as a template
- molecular details similar in all organisms
- General Mechanics – semi-conservative, replication fork, leading strand, lagging strand
- DNA pol only ever adds ntds in 5’ to 3’ direction
- Semi-conservative DNA replication: remember Watson & Crick, Meselson & Stahl
each time a molecule of DNA replicates, the daughter molecules contain one old strand and one new strand of DNA
- Arthur Kornberg (Nobel Prize 1959) were able to identify the requirements for DNA synthesis in vitro (took years)
Required: DNA template, dNTPs (substrates), optimum pH and salt conditions (Mg2+), purified enzyme (called DNA pol l)
- Experiment:
Took E.coli → lysed the cells = created cell free extract
added raiolabelled thymine & ATP as energy source
isolated C14-containing nucleic acid that is digestible by DNase
shows; there is a factor capable of in vitro replication
took 100 lbs of E.coli and lysed them
spun down lysates into subcellular fractions
repeated assay to test if fractions could replicate DNA. Used C14-thymidine to ensure only DNA was replicated (not RNA)
- After a lot of subfractionation and experimentation → Identified DNA polymerase I
- Properties:
5’ → 3’ DNA dependent DNA pol activity = required template & a primer
3’ → 5’ reverse endonuclease activity = proofreading
5’ → 3’ forward endonuclease activity = primer removal
- These activities are seperable. There are distinct domains for each function of DNA pol l
- Was DNA pol I the primary enzyme for DNA synthesis? Nope
Not fast enough compared to DNA synthesis rates seen in vivo
- Had to prove it: John Cairns asked his lab assistant Paula DeLucia to look for a mutant in DNA pol l → found polA mutant
The experiment: take heavily mutagenized E.coli, randomly picked colonies, measured incorporated C14-T like Kornberg
wt sensitive to DNA damage, polA mutants were much more sensitive to DNA damage
Tells us → DNA pol l not primary polymerase (grows but grows slower) & DNA pol l is involved in DNA repair
DNA polymerase ll/ DNA polymerase lll
- DNA Polymerase II was discovered as the prominent DNA polymerizing activity that remained in cells, where polA was mutant - by Thomas Kornberg, the son of Arthur Kornberg, who discovered DNA Pol I
- DNA pol ll can compensate for polA- mutants (used in DNA repair pathways), but still not dominant polymerase
- DNA polymerase: catalyzes phosphodiester bonds in 5’ → 3’ direction
- Substrate are dNTPs → triphosphate (high energy)
2 PO4 are liberated upon polymerization
- Polymerization requires: template, dNTPs, primer (3’OH; all DNA polymerization requires an existing 3’OH)
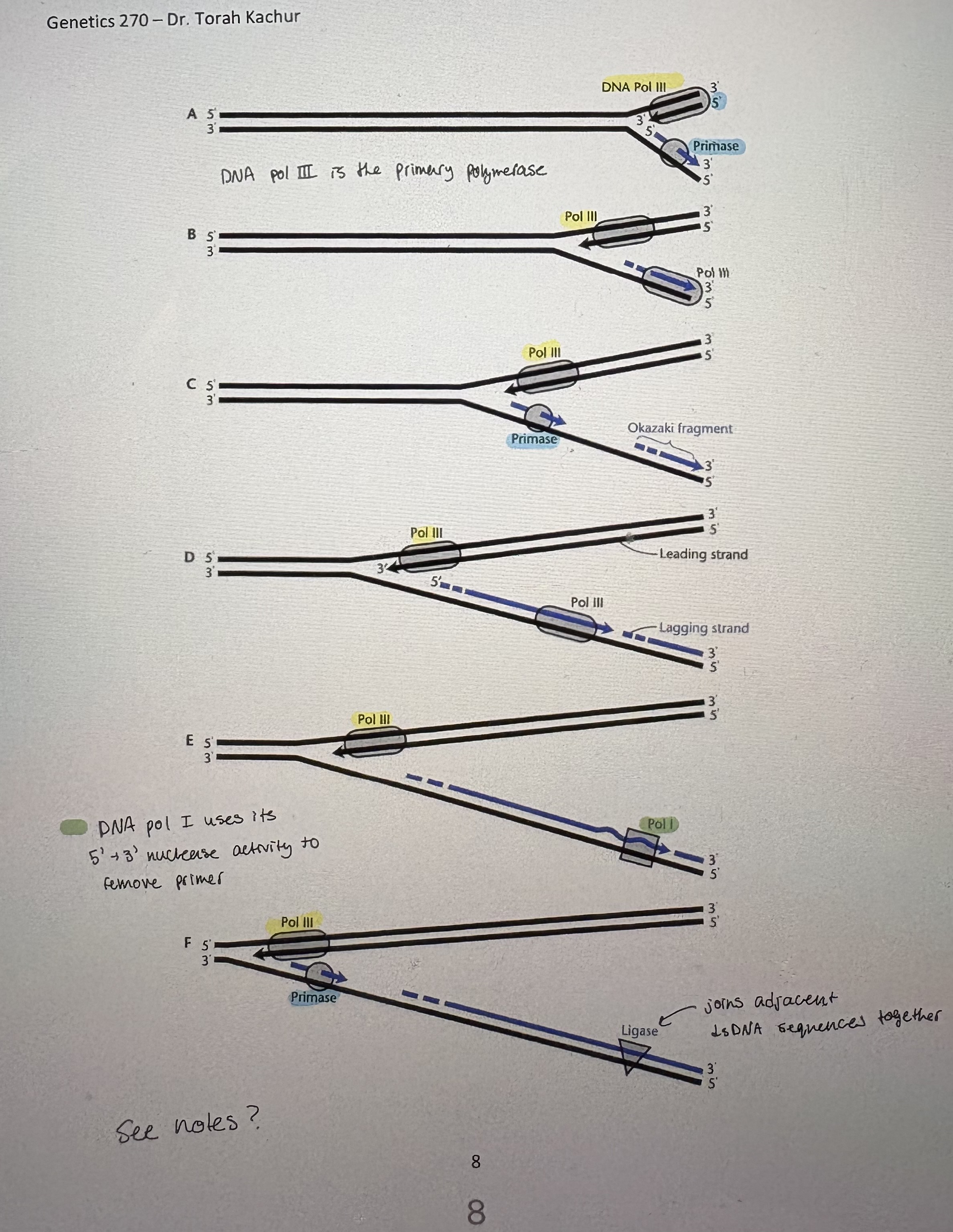
Discontinous replication
- DNA polymerase moves along template in 3’→5’ direction, synthesizing DNA only in a 5’→3’ direction
- Lagging strand: daughter strand that grows in: 5’-3’ direction but is polymerized off strand that runs 5’-3’ away from the origin of replication
synthesized in small fragments in opposite direction of fork mvmt
synthesized as Okazaki fragments
- Leading strand: daughter strand that grows in: 5’-3’ direction and proceeds on the template strand that runs 3’-5’ away from the origin of replication
synthesized in the same direction as fork opening
- BUT = all DNA pol require an existing 3’OH thus need primer → RNA based
- Primase/DnaG (RNA pol): ~10-12 ntds long. Primed every 2 kb (see pic)
primase recognizes 3’GTC5’ & primase after T
Primase is needed to start leading strand and at every 5’ end of Okazaki fragments
- Identification of Okazaki fragments: follow the incorporation of [3H]-thymidine into replicating E. coli DNA
Condition 1: grow bacteria in 3H-thymidine (radiactive ntd) for a brief period. Immediately isolate DNA
Condition 2 (pulse-chase method): grow bacteria in 3H-thymidine (radiactive ntd) for a brief period. Transfer to a non-radioactive medium for several generations
- Used density-gradient centrifugation to distingusih resulting DNA based on molecular mass = radioactive DNA has more mass
Cesium-chloride gradient. Diff mass DNA will appear at diff location sin coloumn.
Condition 1: most radioactivity was found in short fragments. Condition 2: most radioactivity was found in larger fragments
Thus, lagging strand is synthesized in short, discontinous fragments
- Reality? DNA is replicated by a large complex called the replisome. As DNA is seperated by helicase, the lagging strand template loops out & back into replisome (see pic)
Accessory proteins/Models of DNA replication
- DNA replication complex: Form the holoenzyme along with DNA polymerase III (subunits α and ε)
Include: RNase H, β clamp, clamp loaders, editing function
- Involved in:
Processivity; β clamp forms a ring around DNA = holds DNA pol onto DNA
RNA primer removal; RNaseH function of DNA pol l
Proof-reading
- DNA pol lll complex has many genes involved
- most bacterial and phage DNA molecules replicate as: a bidirectional replication fork that forms a theta structure in a circular bacterial genome
- first observed by Cairns: showed that
bacterial DNA is circular, there is a single origin of replication, replication was semi-conservative
- since then, electron micrographs of replicating circular DNA obtained for plasmids, phages, viruses
- Two forms of circular DNA replication
1. θ replication (see pic)
replicating circles resemble Greek letter θ → θ replication
due to BIDIRECTIONAL REPLICATION of circular DNA molecule moving away from origin
ter sites: replication fork can pass through them, but can’t back up. Replication terminates: at a site in between ter sites (in E.coli = terC)
ter sites are bound by tus proteins = prevent DNA pol from passing through 2nd replication fork.
σ replication/rolling circle replication (see pic)
replicating DNA looks like Greek letter σ
used by: some phage w/ circular genomes, some plasmids
Occurs in 2 stages: 1) nick made by Rep protein, recognizes DSO sites (ds origin), uses 3’OH off nick to initiate replication. 2) DNA pol lll carries out replication
Cooridnation of chromosome replication with cell division
- cell division must be coordinated with replication to ensure each of daughter cells receives a chromosome
- Helmstetter & Cooper 1968: measured DNA content of synchronously growing bacteria at different stages of the cell cycle under varying growth conditions:
1) minimal media 2) complete media
cells were attached to a memb thus only newly divided daughter cells could detach into liquid media
fast growth (CM) → ~30min generation time. Slow growth (MM) → ~70min generation time. BUT replication takes ~40min
Rate of DNA replication is controlled by the rate of initiation
- during fast growth replication initiation occurs before previous round is complete
- controlled by levels of DnaA? How do you start dividing before replication is complete
DnaA controls initiation of replication, binds to OriC, needs ATP to bind thus replication is controlled by rate of metabolism
- after (θ) replication, 2 daughter DNAs may still be intertwined or looped
type II topoisomerases: initiate dsDNA cuts to relieve any torsional tension. Reseal DNA cut after unlooping/untangling
Biological indication of DNA damage / Direct repair
- exposure to radiation and chemicals leads to: cell damage, DNA damage → can prevent replication
- measured with SURVIVAL CURVES: expose cells to small doses of potentially DNA damaging agent (intermittent vs continously)
If cells have DNA repair mechanisms, more cells will survive intermittent treatment even if overall mutagenic dose is the same (death is delayed w/DNA repair)
- Survival curves are not linear:
low doses; fraction of cells that survive must have DNA repair that can fix damage
high doses; amount of DNA damage overwhelms repair reponse
- Mechanism of killing by mutagens: blocks in DNA replication
ss breaks = break phosphodiester bond causing a nick.
Mutagens that do not affect base pairing
eg UV light → can induce ssDNA breaks (& dimers). Cannot be replicated nor transcribed thus can trigger programmed cell death
ds breaks
eg Induced by high frequency radiation; gamma rays. Can induce translocations, inversions, deletions
1. Photoreactivation
- UV light induces cyclopyrimidine dimers; covalent bond between adjacent pyrimidines
- pyrimidines can no longer base pair. Also causes a distortion in helix → difficult to replicate across
- Mechanism: DNA replication will throw A’s acroos any lesion in DNA. 1 (-1 deletion) or 2 As (normal)
- UV dimers can be repaired but that repair is light dependent
There is an enzyme that perfectly reverse the mutagenic damage
Photolyase needs light activation (die slower in light)
Photolyase can directly break cyclopyrimidine dimer bond
- The enzyme of repair is PHOTOLYASE: ex of direct repair
Evidence for photolyase: UV irradiated phage, plaqued onto bacteria (in light and in no light), assay for phage growth
Shows → induction of damage isn’t required to trigger repair pathway. Constant DNA damage surveilance
Indirect repair/ Specific repair pathways
- How does the cell deal with damage to DNA when it can’t reverse it directly?
- Shuster (1964) showed repair of UV-lesions without light and without replication to show that DNA repair can occur in relative genome stability. What he did:
Grew thymine-, arginine-, uracil- auxotrophs = can’t replicate
allowed some DNA replication by incubating cells in arginine & uracil
then added 3H-thymine. Measured 3H-thymine incorporation.
Group A = irradiated w/ UV light before replication start. Group B = irridiated 45 min after replication starts
Conclusion: rate of uptake was greater in Group A = evidence for repair pathways independent of replication & light
- Some repair pathways only repair certain types of damage
- Many have overlapping general principles of cutting out the lesion – sometimes carefully, sometimes more aggressively, and then using DNA replication machinery to fill in the resulting gap
- antiparallel and complementary nature of dsDNA means that genetic information has a built in backup system thus complemntary strand can be used as a template for repair
Specific repair pathway; repair of deaminated bases & very short patch repair
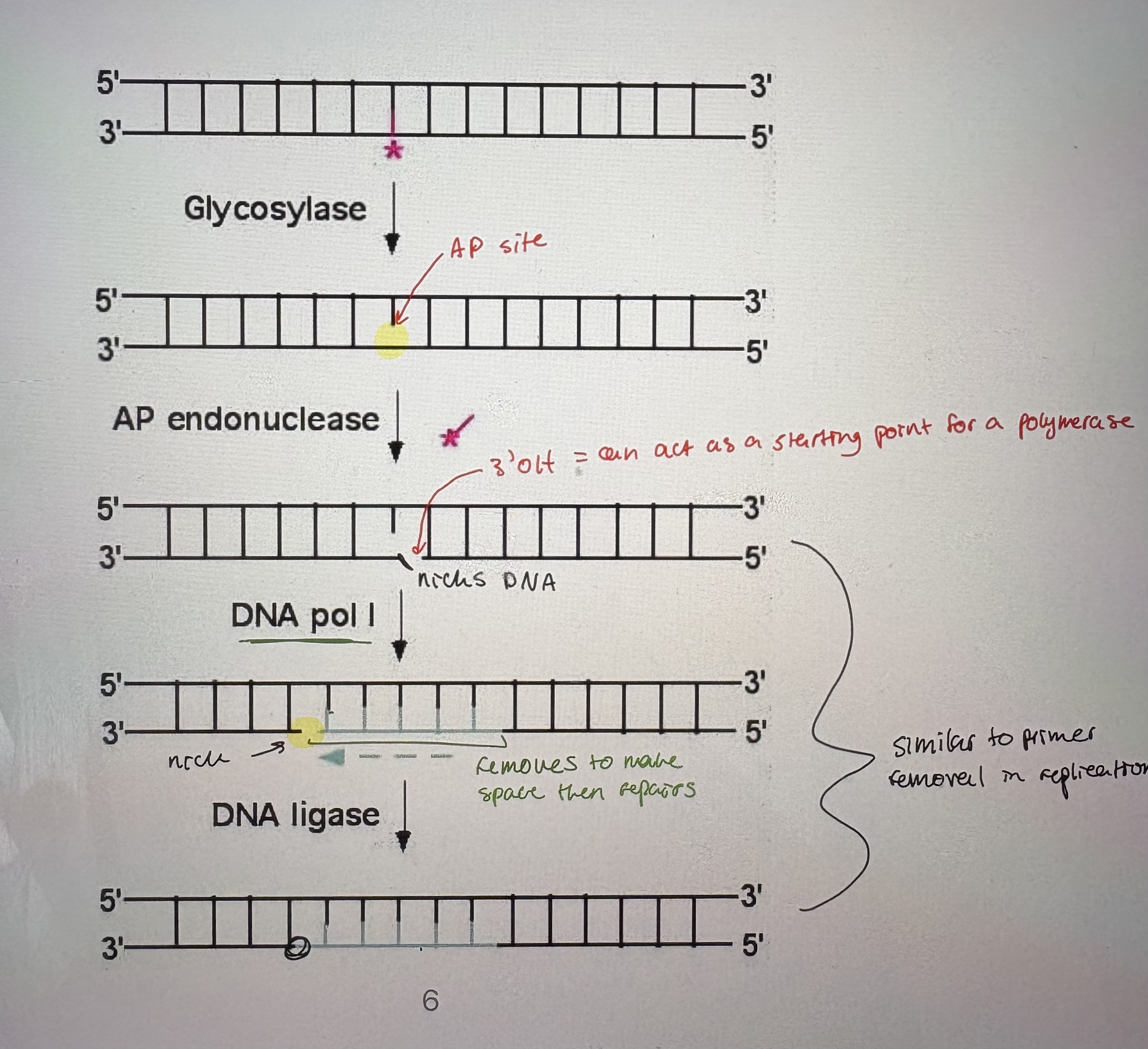
- Repair of deaminated bases occurs via a generic pathway that follows similar steps to many repair pathways using similar enzymes (see pic).
- Glycosolase: break the glycosyl bond between the damaged base and the sugar in the nucleotide
there are many specific glycosylases.
Glycosolases can cleave the base off of the nucleotide. Leaves an AP (apurinic or apyrimidinic) site
- AP endonuclease: Cut sugar-phosphate backbone on 3’Oh side of AP site (endonucleases cleave sugar-phosphate backbone w/in dsDNA molecule).
- DNA pol l: uses 5’-3’ exonuclease activity to remove ntds at nick. Uses its 5’-3’ polymerase activity to fill the gap
- DNA ligase: seals nick at end of newly added DNA. Catalyzes a phosphodiester bond but doesn’t add ntds.
- repairs T:G mismatches that result from deamination of 5-CH3-Cytosine. Very specific.
- 5-CH3-Cystosine: methylated cystoine is common in bacteria because its protects sequences from being cut by its own restriction enzymes
catalyzed by Dcm (methyl transferase)
- not recognized by mismatch repair → occur after replication. Lots of methylates cystoine inc G=T mismatches
Vsr endonuclease: bind to G-T bp but only T is removed
DNA pol l: repairs gap (exonuclease, polymerase)
DNA ligase: seals the nick
- Vsr endonuclease encoded downstream of Dcm methyltransferase (enzymes that methylates C) → ensures cells inhert gene to methylate C & repair common errors that arise from that