Telomeres
1/27
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
28 Terms
How do telomeres protect genome stability? Describe the basic structure of telomeres.
DSB-like structures exist in the genome (chromosome ends) - telomeres protect DSB-like ends from recognition as a DSB by DDR mechanisms
prevents chromosomal translocation and genome instability
Telomeres consist of 2-10kb of TTAGGG repeats, with single stranded 100-200bp overhand strands/
How was the function of TRF1 discovered? What is the main action of the Shelterin complex in telomeres?
Immunofluorescence showed TRF1 protein signals localised at chromosome ends - TRF1 promotes telomere replication
Shelterin blocks activation of DDR through action of 6 components
each domain has individual proposed functions, however not fully understood
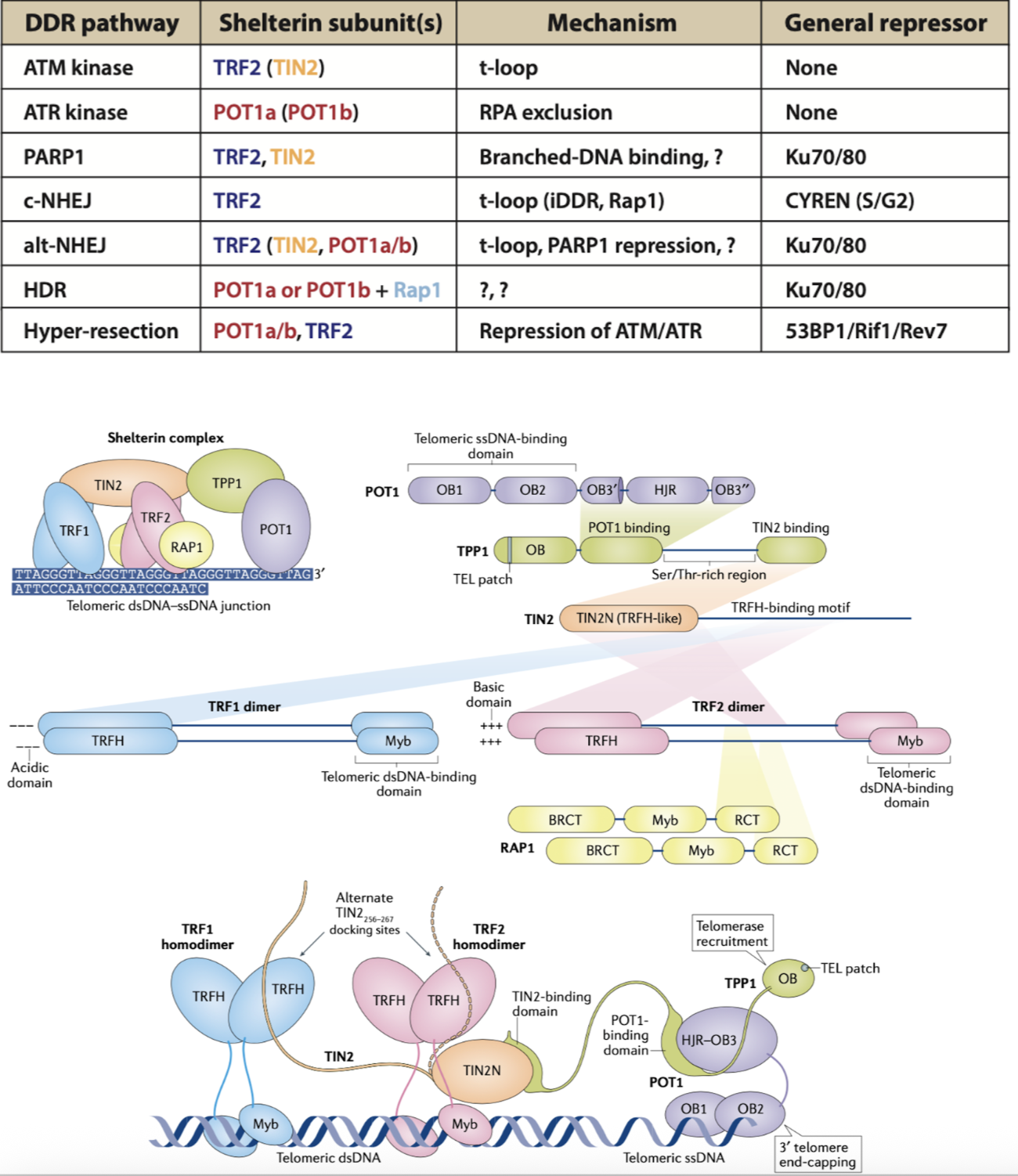
How has KD of TRF2 by CreLox tech improved understanding of the function of TRF2 in telomeres?
Deletion of some Shelterin proteins is lethal (TRF2)
KD of TRF2 by CreLox lead to loss of TRF2 and Rap1 (suggests these proteins mediate eachothers expression)
TRF2 represses ATM kinase and NHEJ at telomeres
KD TRF2 mice express high 53BP1 (DSB has been recognised and NHEJ activated) - TRF2 protects ends
ATM KO reduced 53BP1 expression
TRF2 prevents ATM from inducing repair to telomeres
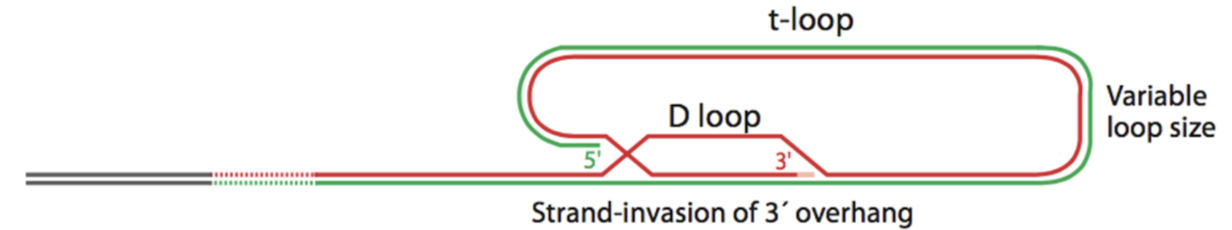
How do T loop structure protect telomeres?
Loop structures at the ends of telomeres were discovered in 1999.
ssDNA overhand strand invades dsDNA, displacing one strand of dsDNA > D-loop and T-loop - hides DNA ends from repair enzymes
TRF2 KO lost T-loops
How does the POT1 Shelterin subunit protect telomeres? Why is HR also repressed in telomeres?
ssDNA binding protein - suppresses ATR kinase by binding RPA in place (prevents activation of DDR)
HR is also suppressed in telomeres - presence of overhang makes telomere length uneven - HR crossing over would form unequal dsDNA - very unstable, this mechanism is inhibited in telomeres
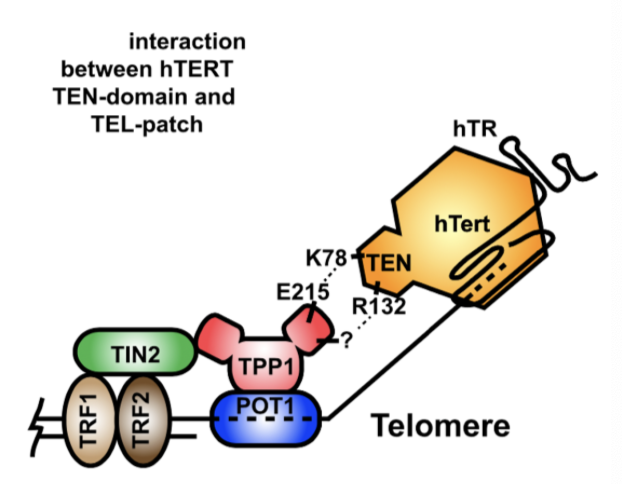
How is telomerase recruited to the telomere?
Telomerase is regulated at multiple levels, expression, biogenesis and at individual telomeres.
Sheltering subunit TERT contains TEN domain, TPP1 (telomerase) contains TEL patch - these domains interact to recruit telomerase to the telomere
Loss of/defective Shelterin at telomeres prevents recruitment and activation of telomerase - CST terminates telomerase action
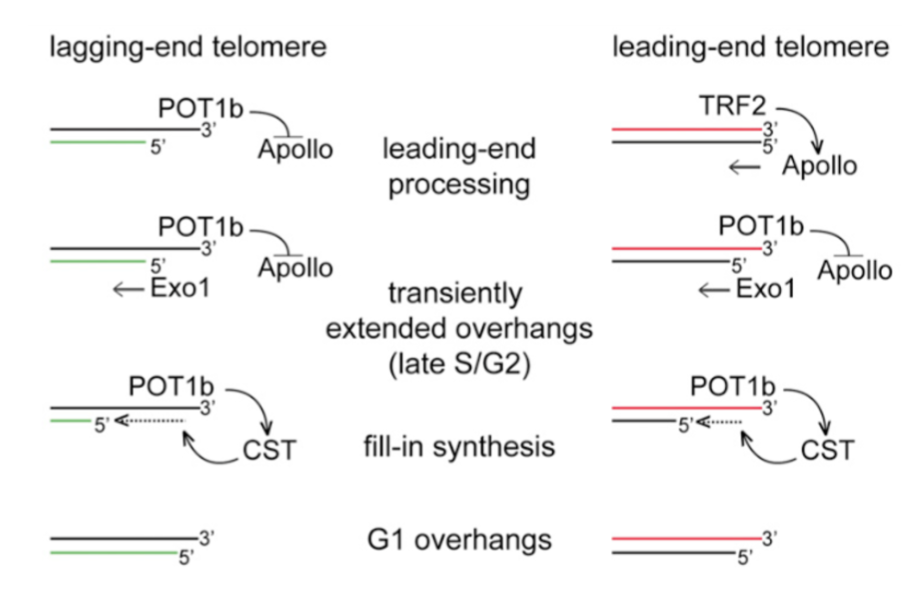
How are G-overhangs generated?
Lagging telomeres generates overhang after replication, POT1b represses Apollo nuclease at the lagging telomere - Exo1 extends overhang
Leading telomere generates blunt ends after replication > no overhang. TRF2 recruits Apollo nuclease at leading telomere to generate overhand - Exo1 extends overhang
in both, CST associates with RPA ssDNA, synthesising the C-strand by recruiting POLa - negatively regulating telomerase and reducing overhang
How does TRF1 control telomerase? Describe the protein counting model for telomerase regulation by TRF1 binding.
TRF1 inhibits the action of telomerase at telomere ends - over expression of TRF1 reduces telomere length, TRF1 loss increases telomere length (per population doubling event)
Protein counting model - all theoretical
Longer telomeres bind more TRF1 - promoting telomerase inhibition and telomere shortening
Shorter telomeres bind lower levels of TRF1 to promote telomerase activity
telomere lengthening and shortening is dynamic
Control of telomerase activity is linked with a window of opportunity for telomerase action - longer telomeres have shorter windows and closed conformations
What are the three types of bulk telomeric DNA? Why is replication stalling in telomeric DNA particularly problematic?
Non-overhang regions (bulk telomeric DNA)
G-quartets
T-loops
TERRAs
Stalled replication forks at telomeres are particularly problematic as they cannot be rescues by convergent fork rescue (as seen in intrachromasomal stalling)
How do G-quartets (G4 DNA) form? What is the issue with these structures in bulk telomeric DNA?
TTAGGG repeats forming 4 stranded DNA structures where 4 guanines pair in one plane - 3 guanine rich planes stack on top of one another to form G-quartets
G-quartets stall replication forks - TRF1 recruits BLM (Bloom helices) to rescue these forks
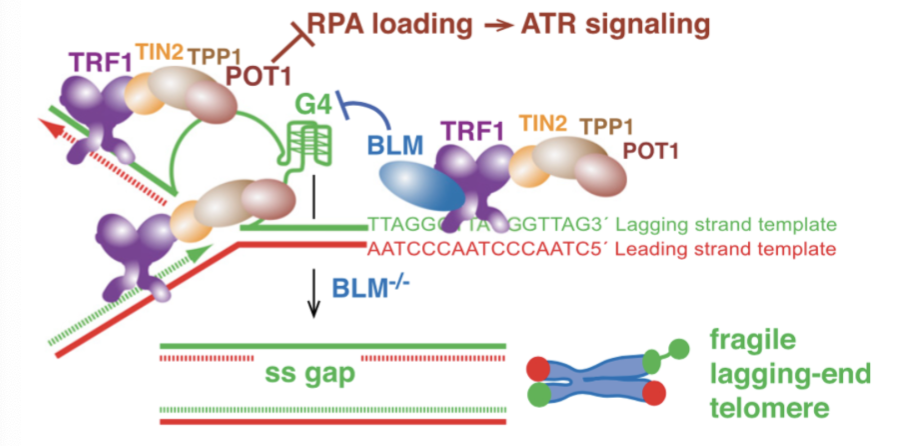
How do TERRAs protect chromosome ends? How does dysregulated TERRA result in telomere instability?
Telomeric DNA is transcribed into RNA which can affect telomeric chromatin - this can leading to favouring rescue of stalled forks by:
regulating telomere length
interacting with TRF1/2
Binds hnRNP1 - promotes RPA-POT1 switch > R-loop formation - RNA displaces one of two DNA strands in dsDNA
unscheduled association with TERRA activates DDR at telomeres
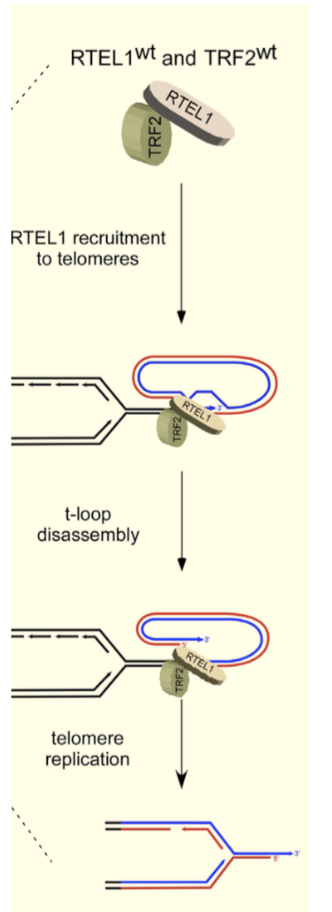
How does RTEL1 and TRF2 control T loop formation and stability?
Outside of S phase, TRF2 is phosphorylated at Ser365 by CyclinA-CDK2, preventing RTEL1 binding
In S phase, phosphorylation is temporarily reversed by PP6R3 phosphatase, allowing RTEL1 helices binding and T-loop disassembly
Rephosphorylation can release RTEL1, protecting T loops from unwinding and inappropriate ATM activation
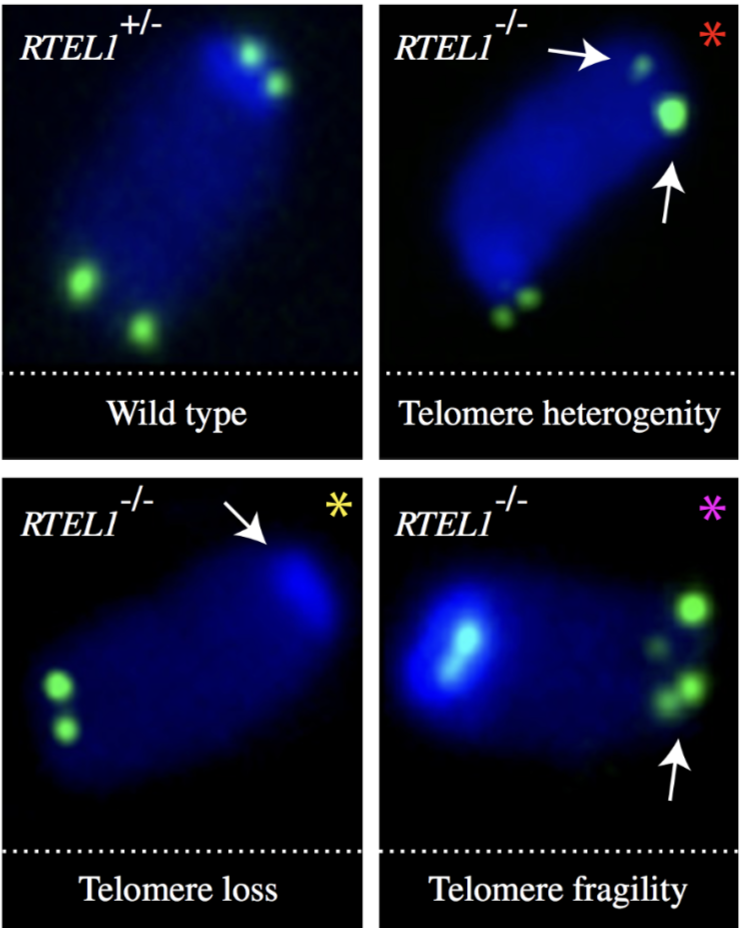
How does RTEL1 dysfunction affect telomeres? How can this be identified in FISH?
RTEL1 mutations prevent TRF2 interaction, resulting in defective replication of telomeric DNA > telomere loss, gaps or fragility due to T loop excision (Hoyeraal-Hreidarsson syndrome)
this can be identified by double signals in telomere FISH of metaphase chromosomes
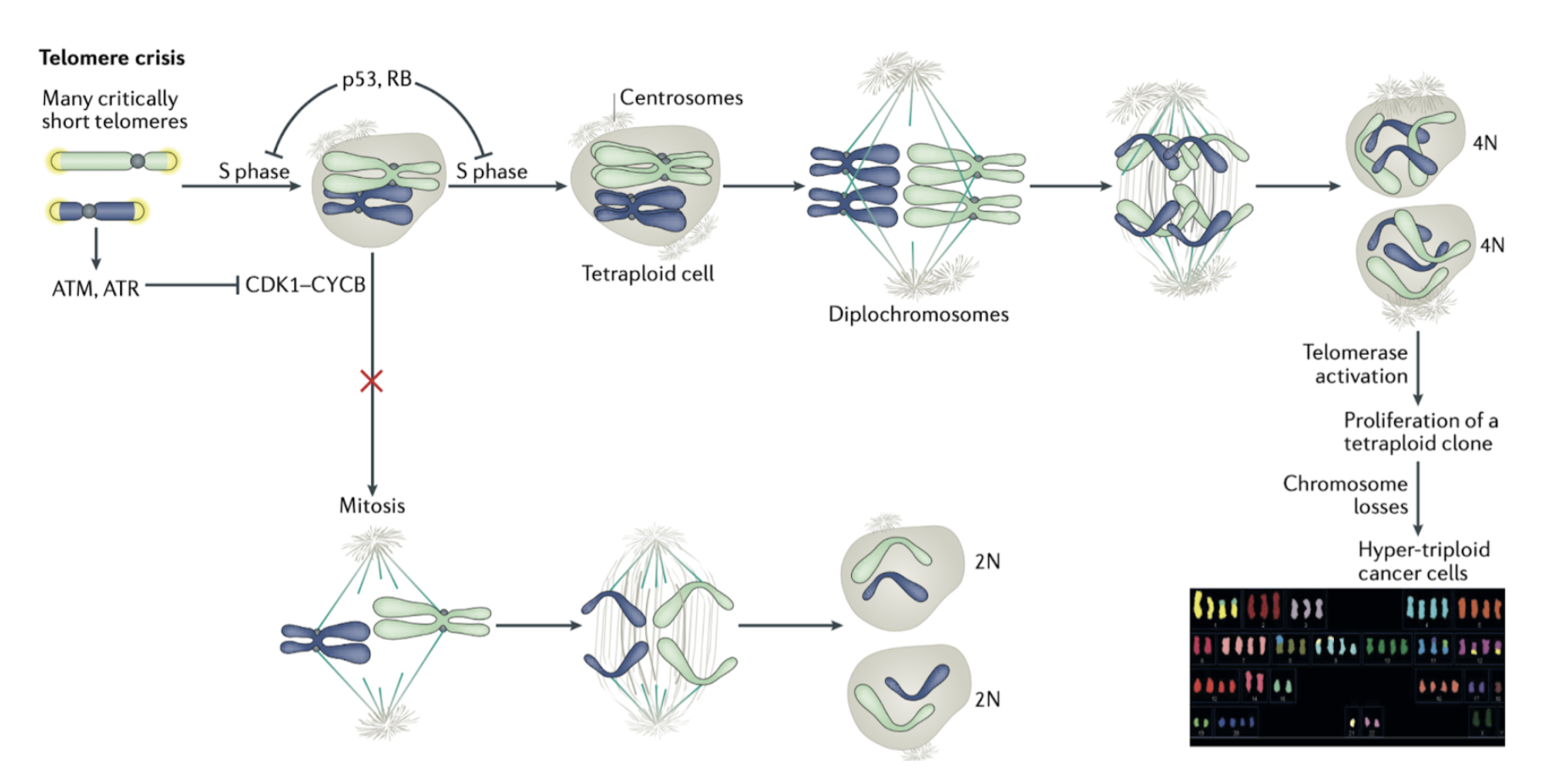
How does telomere crisis promote cancer formation?
Telomere crisis can lead to persistent DDR signalling
eg. ATM/ATR results in prolonged CDK1-CyclinB inhibition > blocked entry to mitosis
Cells can overcome this blockade by bypassing requirements of mitosis > undergo a second S phase
this results in tetraploidy (diplochromosomes) due to double DNA replication and no mitosis > frequently observed in cancer
How does telomeric dysfunction result in chromothripsis and kataegis?
Cancer cells exhibit chromothripsis - chromosome undergoes catastrophic chromosome breakage (fully fragmented), rearranged by NHEJ
Kataegisis is a hypermutation patter of clustered C>T and C>G mutations at TpC dinucleotides - result from APOBEC activity
both due to telomeric dysfunction > fused chromosomes
How does cell death occur in cells with telomeric dysfunction? How was it shown that sencence is actually tumour suppressing/protective?
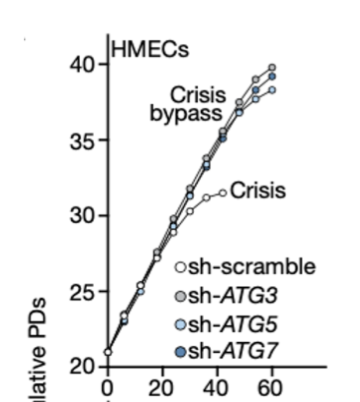
Autophagy related proteins (Atg) coordinate cell death by autophagy at the lysosome
Telomere dysfunction leads to cell death via autophagy
RNAi inhibition of ATG proteins showed bypassing of crisis phase (senescence, end replication problem)
this would eventually lead to unstable, anueoploid genomes - senescence has a tumour suppressive effect
Describe the clinical features of Dyskeratosis congenita, and how it exhibits genetic anticipation.
Dyskeratosis congenita (DC)
Skin abnormalities, BM failure and cancer
defective telomeric maintenance - mutations in genes required for telomerase (Dyskerin)
shorter telomeres and crisis at earlier age
Shows genetic anticipation - worsens across generation as progeny receive shorter telomeres in germline
Give examples of telomere biology disorders.
Pulmonary fibrosis
short telomeres and mutations in telomerase associated with idiopathic pulmonary fibrosis (IPF), mean onset 51 years - can lead to DC in progeny
Aplastic anaemia
clusters with IPF in some families, linked to shorter telomeres and telomerase mutations (3-5% carry TERT/TER mutations)
Hoyeraal-Hreidersson Syndrome (HHS)
Severe form of DC with addition of IUGR, cerebellar hypoplasia and microcephaly
telomeres severely short at 5 years
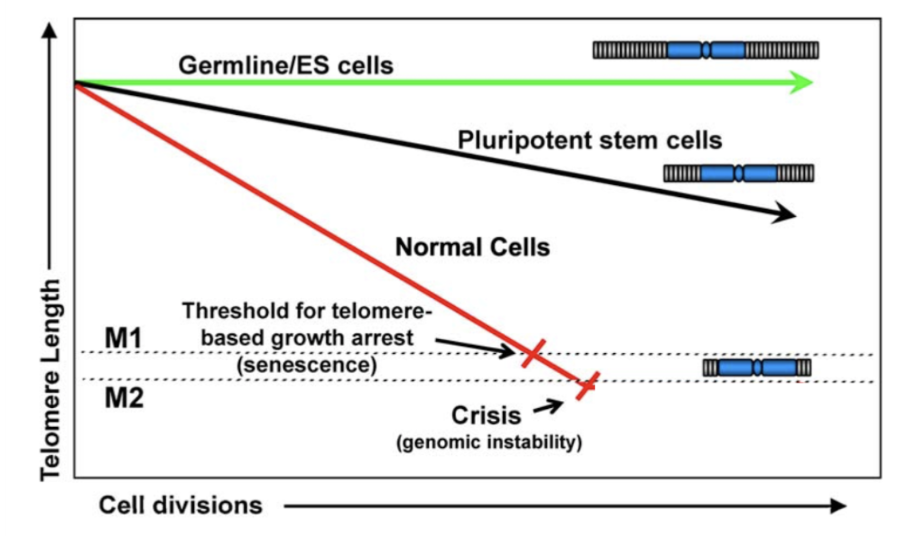
How does telomere shortening induce mortality phases?
Mortality phase 1 (M1) is senescence, enforced by p53 and Rb.
In M1, few cells reach critically short telomere length and enter G1 permanent arrest
Motality phase 2 (M2) is crisis, occurs when senescence is bypassed
many telomeres are critically short, autophagy activates cell feath
Deficient autophagy proteins can lead to M3 mortality phase, even high cell numbers accumulate
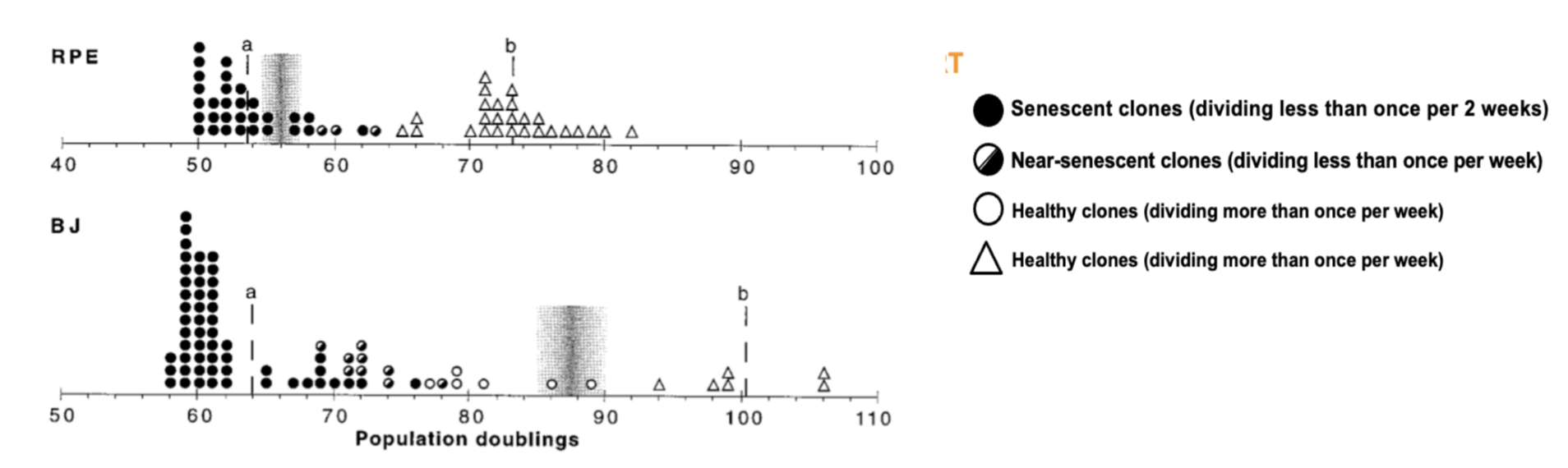
How do cancer cells bypass senescence?
TERT (telomerase) is repressed in most human somatic cells, its ectopic expression is sufficient to immortalise cells - this is often exploited in cancer, allowing cells to bypass Hayflicks limit.
RPE and BJ are human fibroblast cell lines - induced TERT expression rescues senescent cells
How is telomerase reactivated in cancer? What is the two-step model explaining why some tissues exhibit these mutations more than others?
Point mutations reactivate the TERT promoter - initially identified in melanoma (TERT promoter mutations in melanoma (TPMs))
C250T and C228T in 5’UTR ETS factor binding sites of TERT promoter promote telomerase activity
TPMs are most common in cancers originating from tissues with low-self renewal - two step model:
tissue with low self-renewal have full repressed TERT promoters, mutations are required, whereas tissues with renewal potential have low telomerase expression to begin, therefore only needs to be increased by epigenetic modification
How does telomerase up regulation lead to genomic instability in subsequent cell divisions?
Phase1: TPMs arise in early tumourigenesis, upregulating telomerase activity
Phase2: in subsequent divisions, when crucial short telomeres arise, telomerase activity is limited, driving genomic instability
How does Myc amplification promote telomerase activity?
Amplification of MycN activates TERT
in hepatocellular carcinoma, HBV can integrate its genome in close proximity to TERT promoter, placing viral enhancer elements near the promoter
several viral proteins have the capacity to perform this
Hyper-methylation of TERT promoter is the most common activator of TERT in human cancer
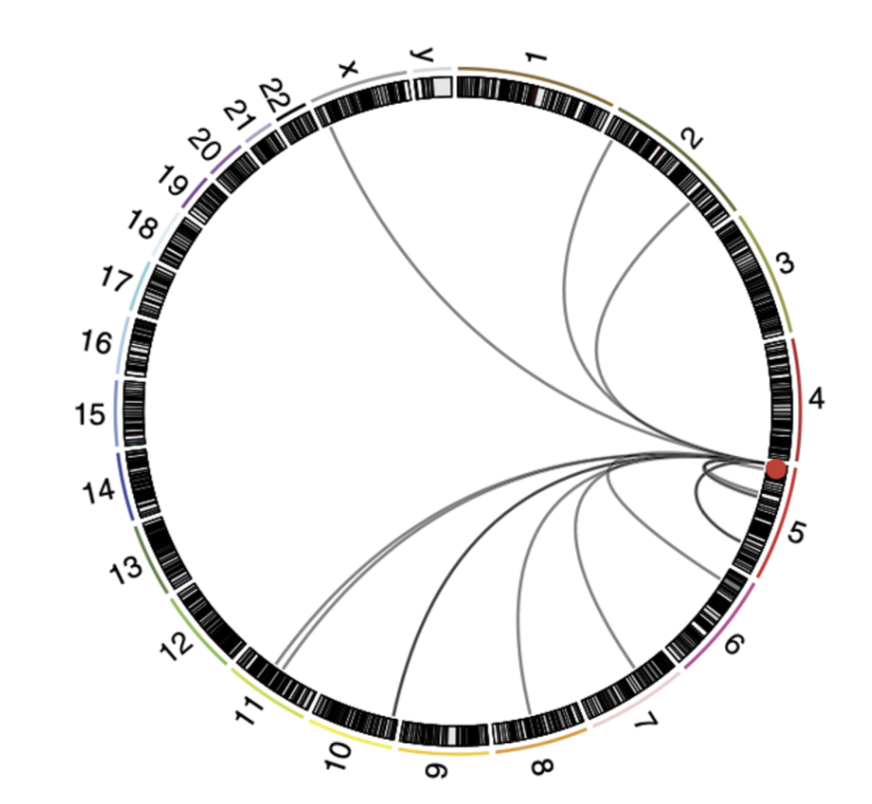
Give examples of chromosomal rearrangements which activate the TERT promoter.
Map of chromosomal rearrangements involving TERT locus (red spot)
TERT, ATRX and MycN mutations are mutually exclusive, suggesting they eat promote individual pro-tumourigneic mechanisms
all correlated with high-risk cancer and poor prognosis
What is the ALT pathway? How can cells with active ALT be characterised?
ALT pathway is activated when TERT activation is not exploited in cancer.
Cells with active ALT are characterised by:
heterogenous telomere length
co-localisation of telomeres with PML bodies in APBs
presence of extrachromosomal telomeric repeats (ECTRs) including circles of partially double-stranded C-rich telomeric DNA (C-circles)
frequent exchanges between sister telomeres (T-SCEs)
How do ATRX and DAXX promote aberrant chromatin decompaction in telomeric DNA?
Associated with mutations in ATRX and DAXX - these act in tandem to form a histone variant (H3.3) - influences nucelosome assembly at repeptitive heterochromatic regions of the genome, including telomeres.
TERT and ATRX/DAXX mutations are mutually exclusive, suggests one mechanism is active in each cancer
~22% of ALT cells do not carry ATRX/DAXX mutations - another gene involved.
In fork stalling:
chromatin alterations at the telomere lead to progressive activate of DDR and telomeric fork stalling
unprepared stalled forks provide a template for recombination between telomeres
What is the theory associating long telomeres with cancer risk?
Long telomeres are more associated with TPMs, and germline heterozygous mutations in POT1, TPP1 and RAP1.
GWAS identified elongated telomeres associated with higher cancer risk - longer telomeres allow a longer window for cancer promoting mutations
supports theory that senescence is protective and tumour suppressing
Why has telomere maintenance evolved in large-bodied, long-lived animals specifically?
Larger animals = more somatic cells = higher mutation potential = higher cancer risk
larger animals must evolve more efficient tumour suppression
Eg. evolution of the repression of telomerase induction of replicative senescence.