T3 - IE2 - Cardiology - Kaur - Anti-arrhythmic Drugs
1/55
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
56 Terms
Class I Sodium Channel Blockers
Quinidine
Procainamide
Disopyramide
Lidocaine
Mexiletine
Flecainide
Propafenone
Class IA anti arrhythmic drugs
Quinidine
Procainamide
Disopyramide
Quinidine
-prototype drug for class IA
-alkaloid found in Cinchona bark
-diastereomer of quinine
-quinoline ring and bicyclic quinuclidine ring system w/ hydroxymethylene bridge connecting them
-can be given IV as the gluconate salt
-good oral absorption of salt forms of quinidine (95%)
Quinidine structure
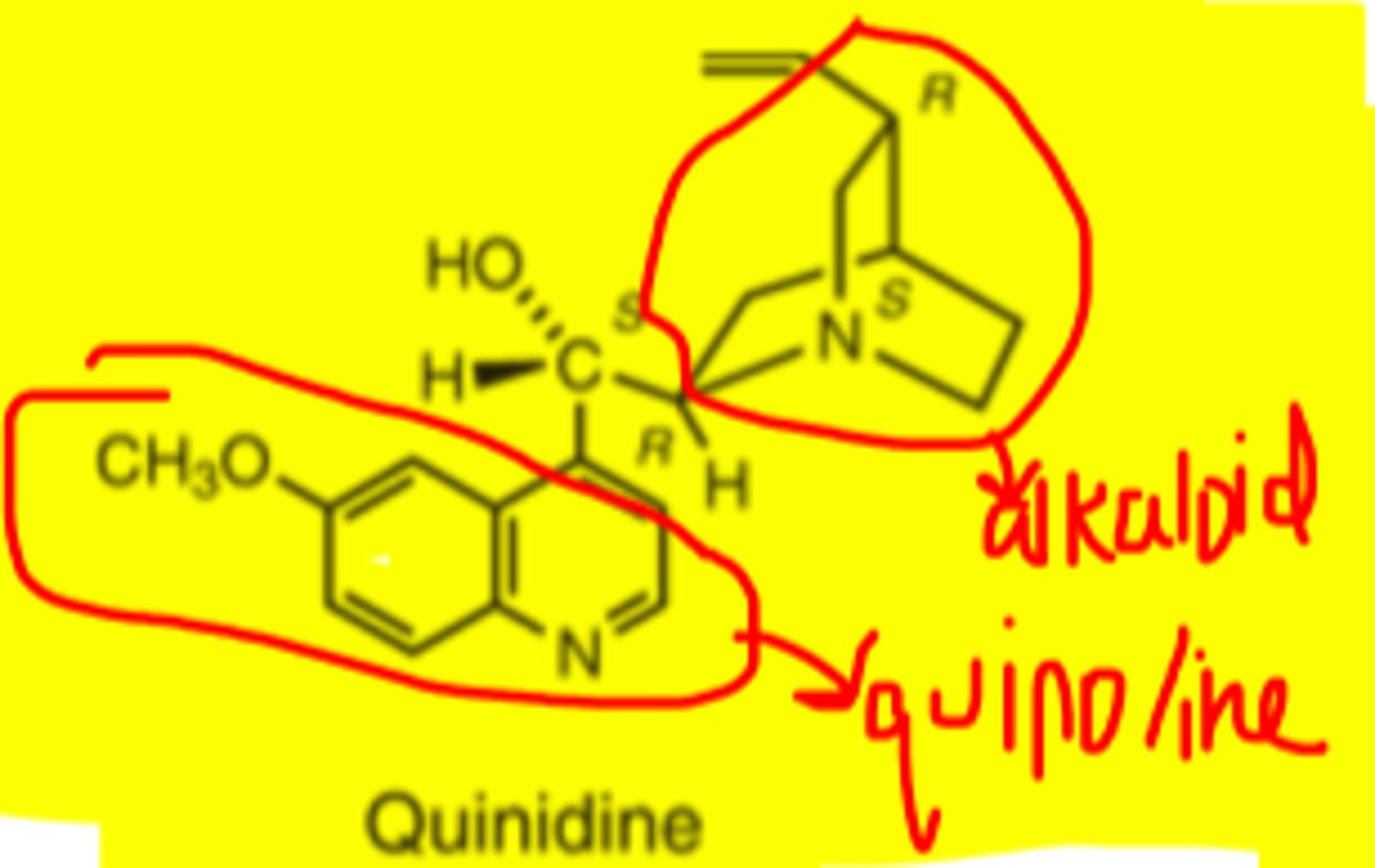
Because of the basic character, quinidine is always used as __________ __________ _________ _________
water soluble salt form
ex: sulfate, gluconate
Metabolites of quinidine
-hydroxylated derivatives at either quinoline ring, or at the quinuclidine ring
-metabolites have only 1/3 activity of quinidine
Digoxin-quinidine interaction
-quinidine (P-gp substrate) competitively inhibits renal tubular secretion of digoxin via P-gp efflux pump
-reduces digoxin renal secretion by 60%
-increases plasma conc. of digoxin to toxic lvls
Procainamide
-can be effective in pts unresponsive to quinidine
-peak plasma levels 45-90 mins after oral administration
-70%-80% of dose is bioavailable
Procainamide structure
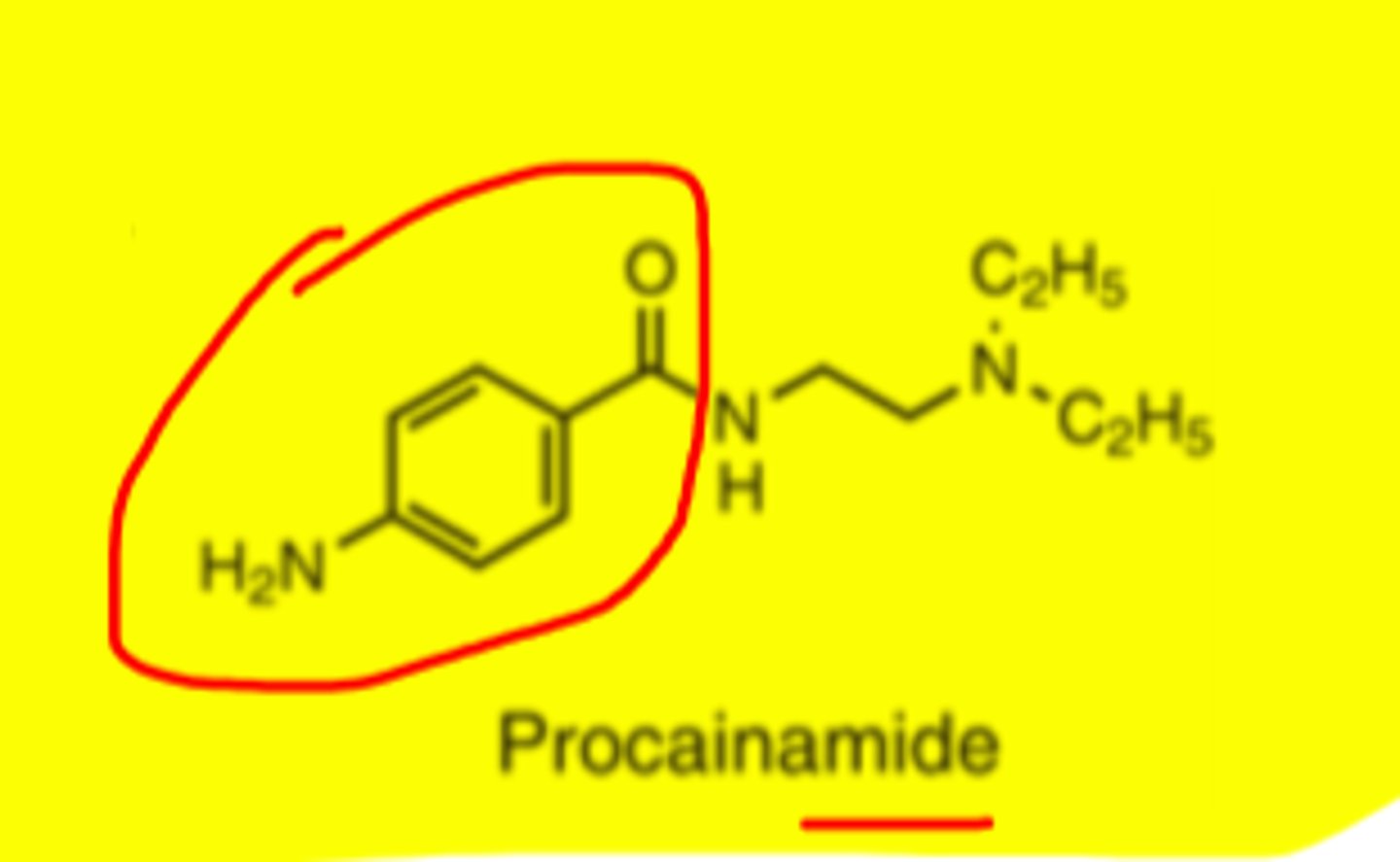
Metabolites of procainamide
p-aminobenzoic acid and N-acetylprocainamide
(less common metabolites)
The acetylated metabolite of procainamide is also active as an _____________.
Antiarrhythmic
-formation accounts for up to 1/2 of administered dose and catalyzed by N-acetyltransferase
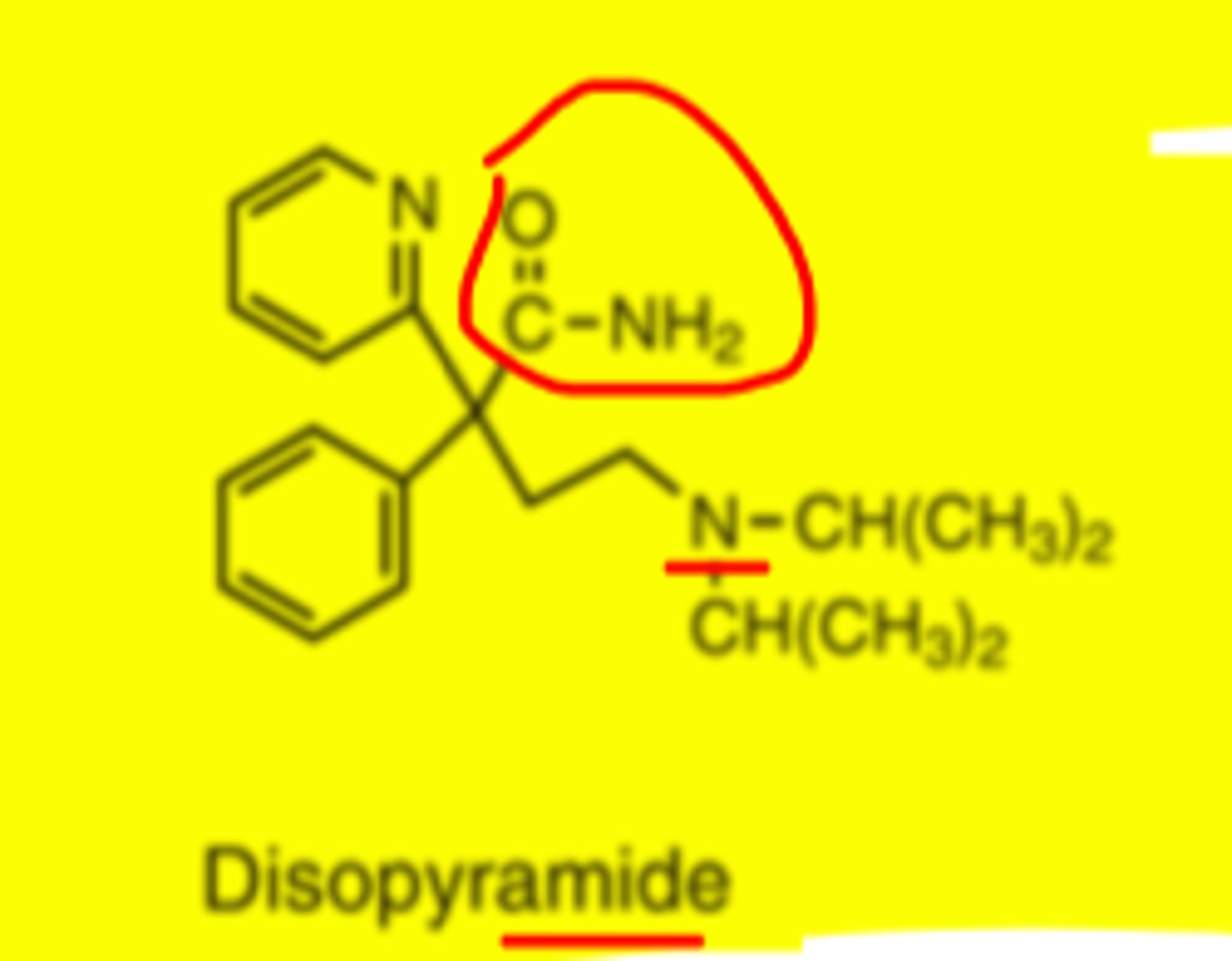
Disopyramide
-used orally to treat ventricular and atrial arrhythmias (well behaved drug)
-structural dissimilarity to procainamide, but similar cardiac effects
-rapidly & completely absorbed from GI tract
-peak plasma levels 1-3hrs
-plasma half life 5-7 hrs
Disopyramide metabolites
-N-dealkylated metabolite by CYP3A4
-retains approx 1/2 of anti arrhythmic activity
Disopyramide structure
Phase I and II metabolism
-Phase 1: P450 hydroxylation to make molecule more reactive, polar, and hydrophilic (phenol)
-Phase 2: glucuronide conjugation to increase charge and polarity (glucuonide)
Class IB anti arrhythmic drugs
Lidocaine
Mexiletine
Lidocaine
-drug of choice for emergency tx of ventricular arrhythmias
-rapid onset of anti arrhythmic effects on IV
-therapy can be rapidly modified
-effective as anti arrhythmic only when given parenterally (IV route most common)
-rapid.hepatic metabolism
-plasma half life 15-30mins
Metabolites of lidocaine
N-deethylation, followed by amidase-catalyzed hydrolysis into N-ethylglycine and 2,6-dimethylaniline
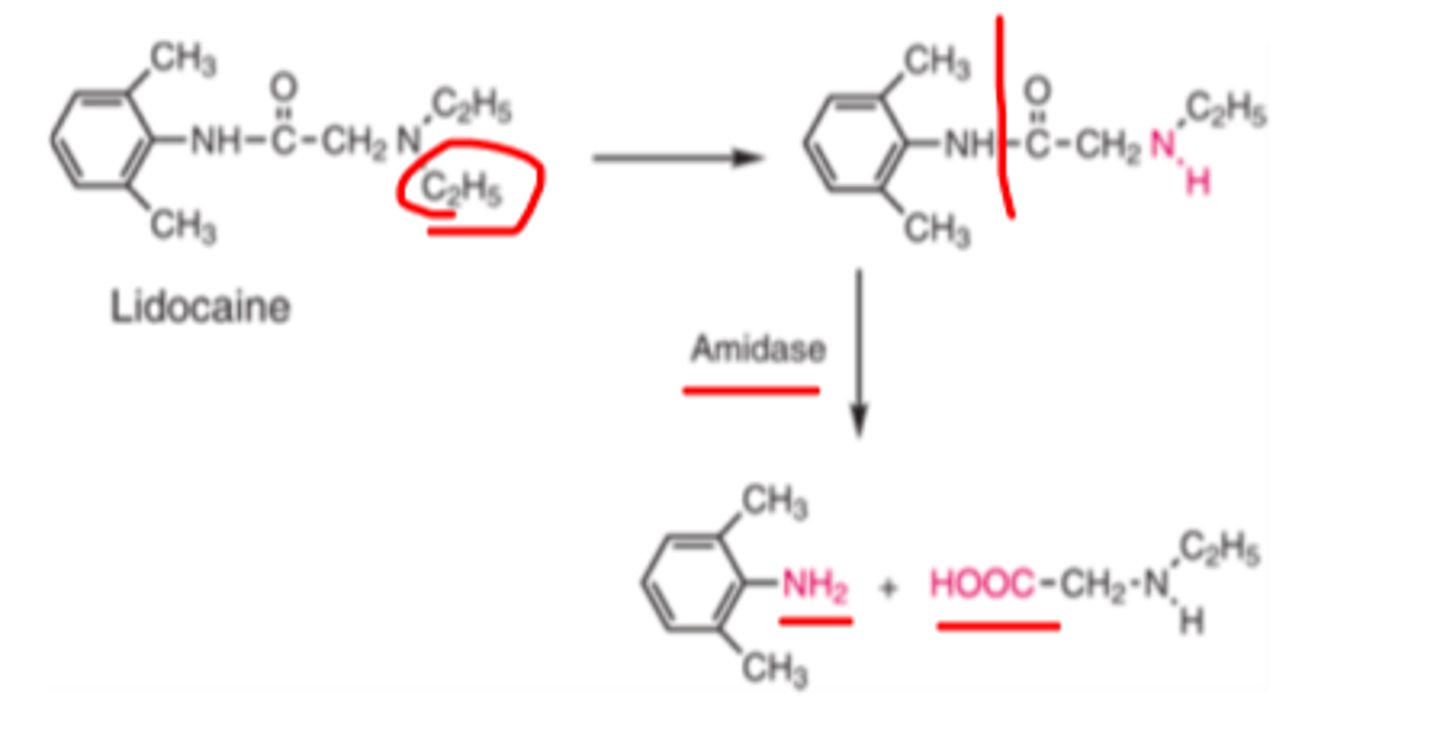
Lidocaine structure
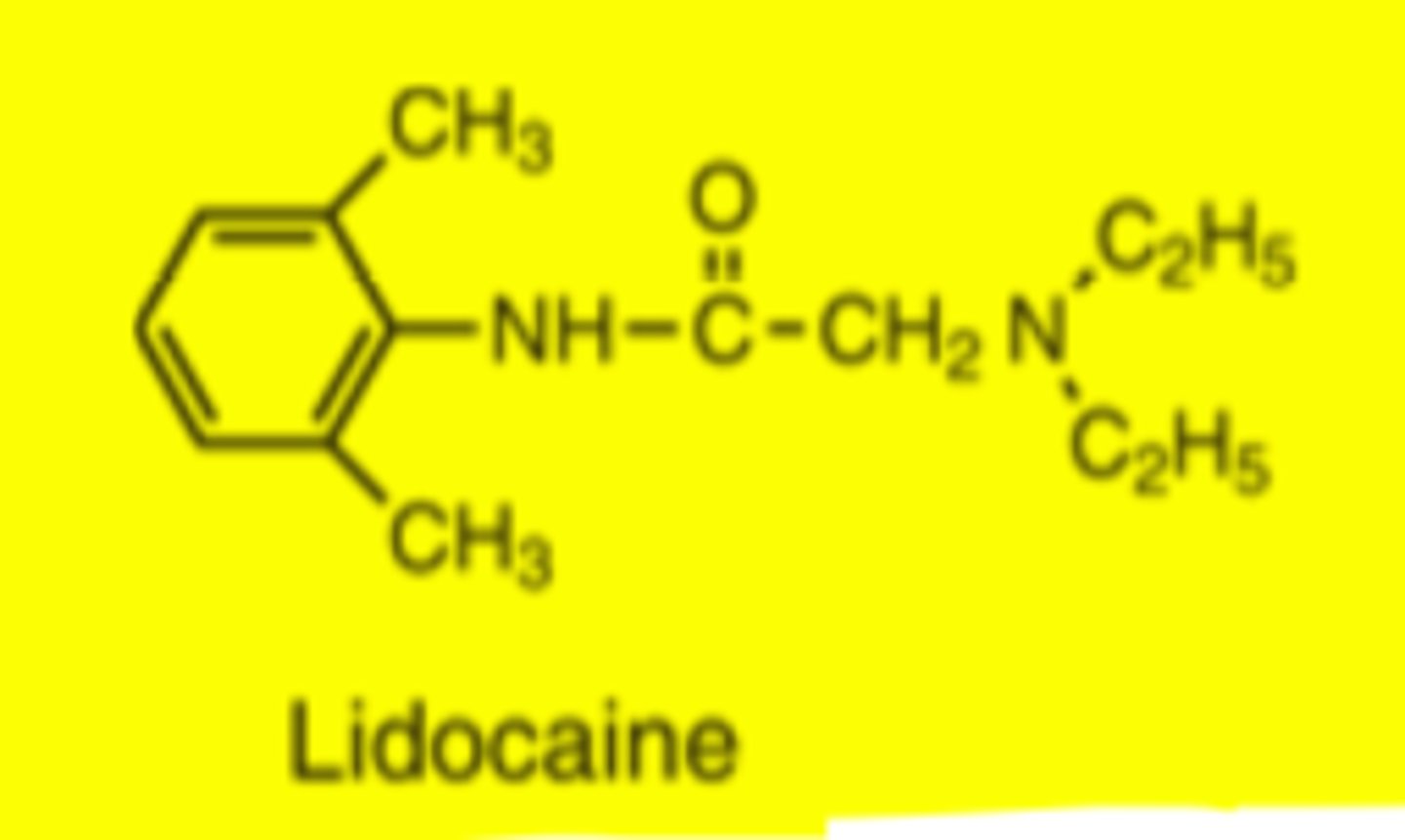
Mexiletine
-similar to lidocaine in effects and therapeutic
-treats and prevents ventricular arrhythmia
-very good oral activity and absorption
-long plasma half life (12-16 hrs)
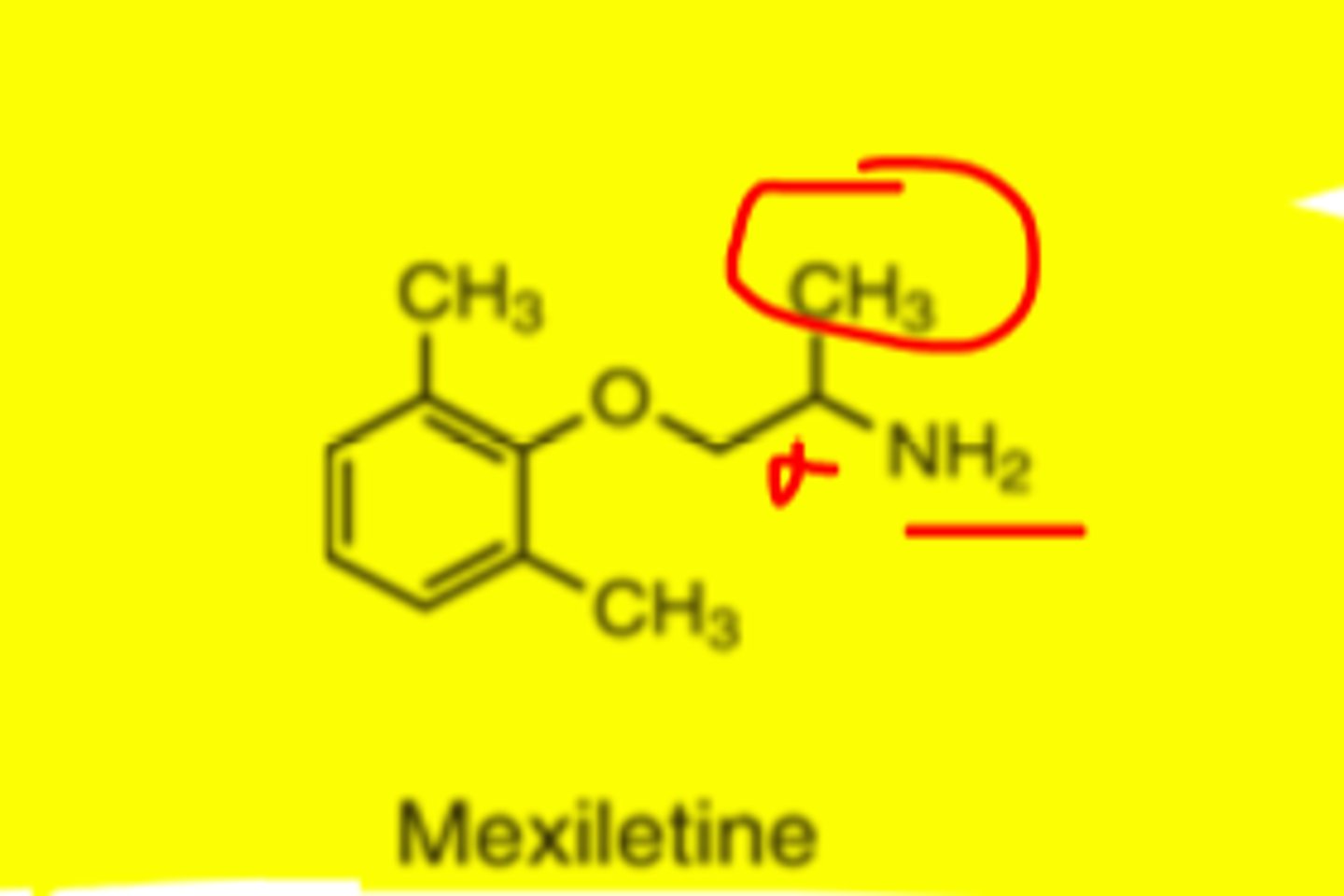
Mexiletine structure
alpha methyl group slows rate of metabolism and contributes to oral activity
Class IC anti arrhythmic drugs
Flecainide
Propafenone
Flecainide
-fluorinated benzamide derivative
-available as acetate salt
-oral form well absorbed
-plasma half life 14 hrs
Flecainide structure
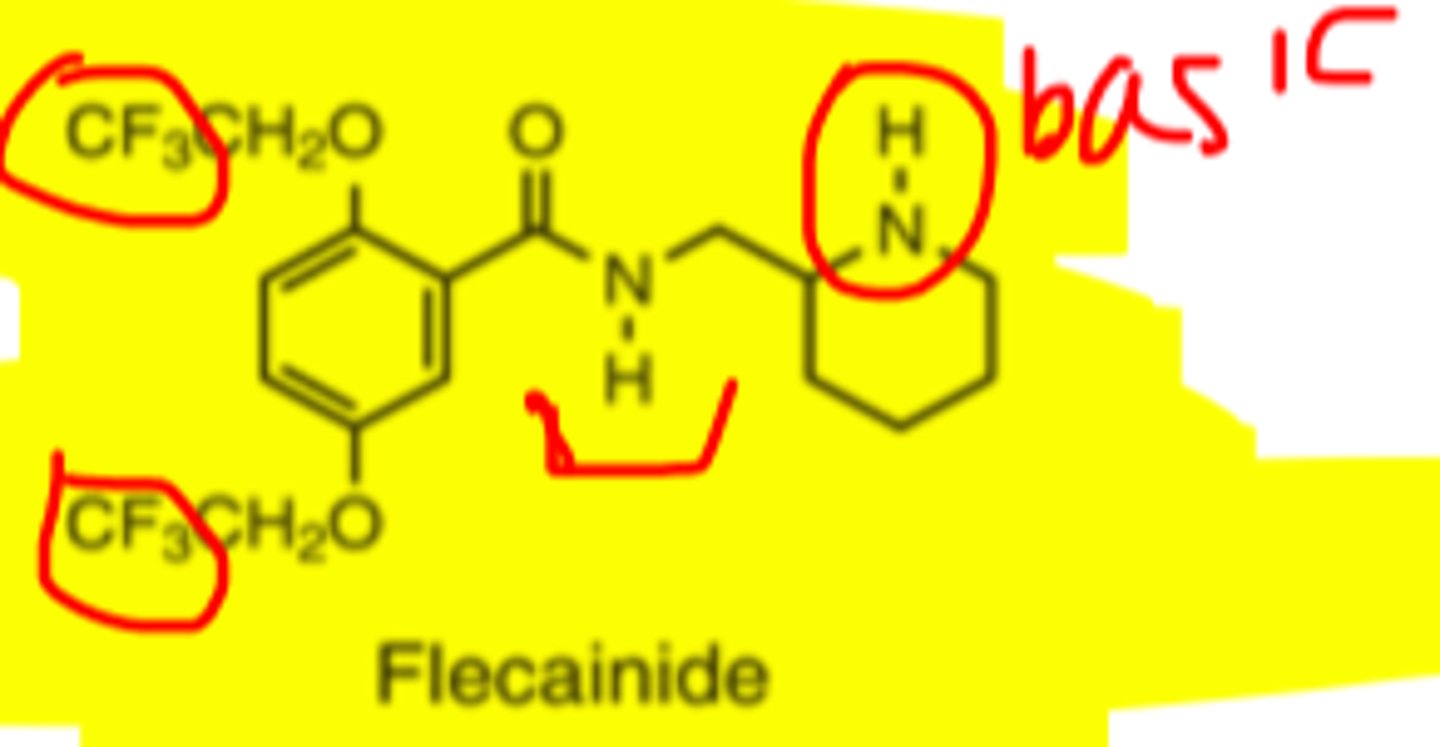
Flecainide metabolism
-1/2 oral dose is CYP2D6 metabolized in liver
-1/3 excreted unchanged in urine
Propafenone
-structurally related to IC aa's and B-blockers
-oral and IV, parenteral not in US
-rapidly & almost completely absorbed from GI tract after oral administration
Propafenone metabolism
-hepatic CYP2D6 enzymes
-90% of pts, rapidly & extensively metabolized (elimination half life: 2-10 hrs)
-slow metabolizers elimination half life: 10-32 hrs
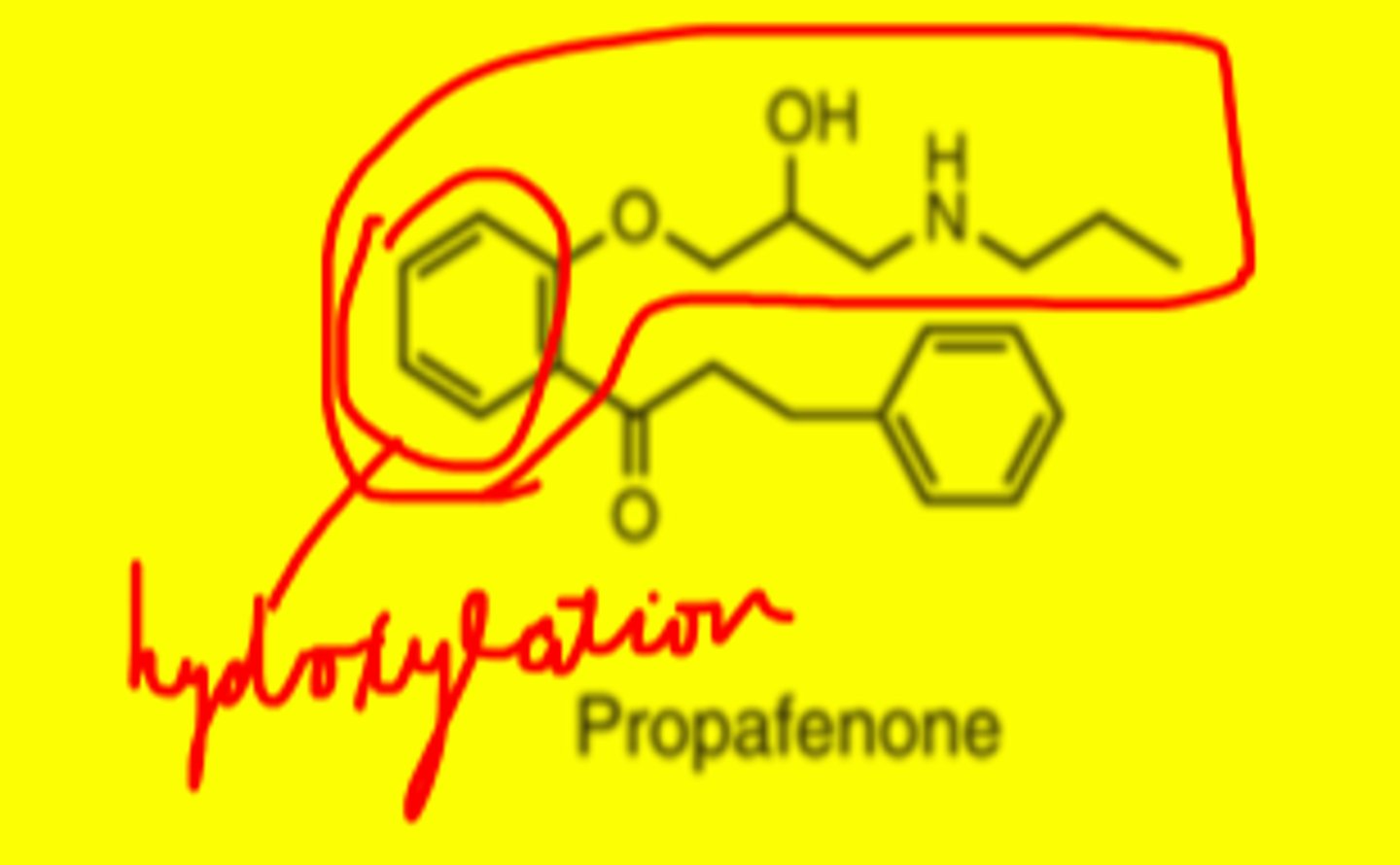
Propafenone structure
-phenyloxypropanolamine pharmacophore
-phenyl ring gets hydroxylated by CYP2D6
Class II Antiarrhythmic Drugs
B-adrenergic receptor blockers
ex:
propranolol (phenyloxypropanolamine)
sotalol (methanesulfonanilide derivative)
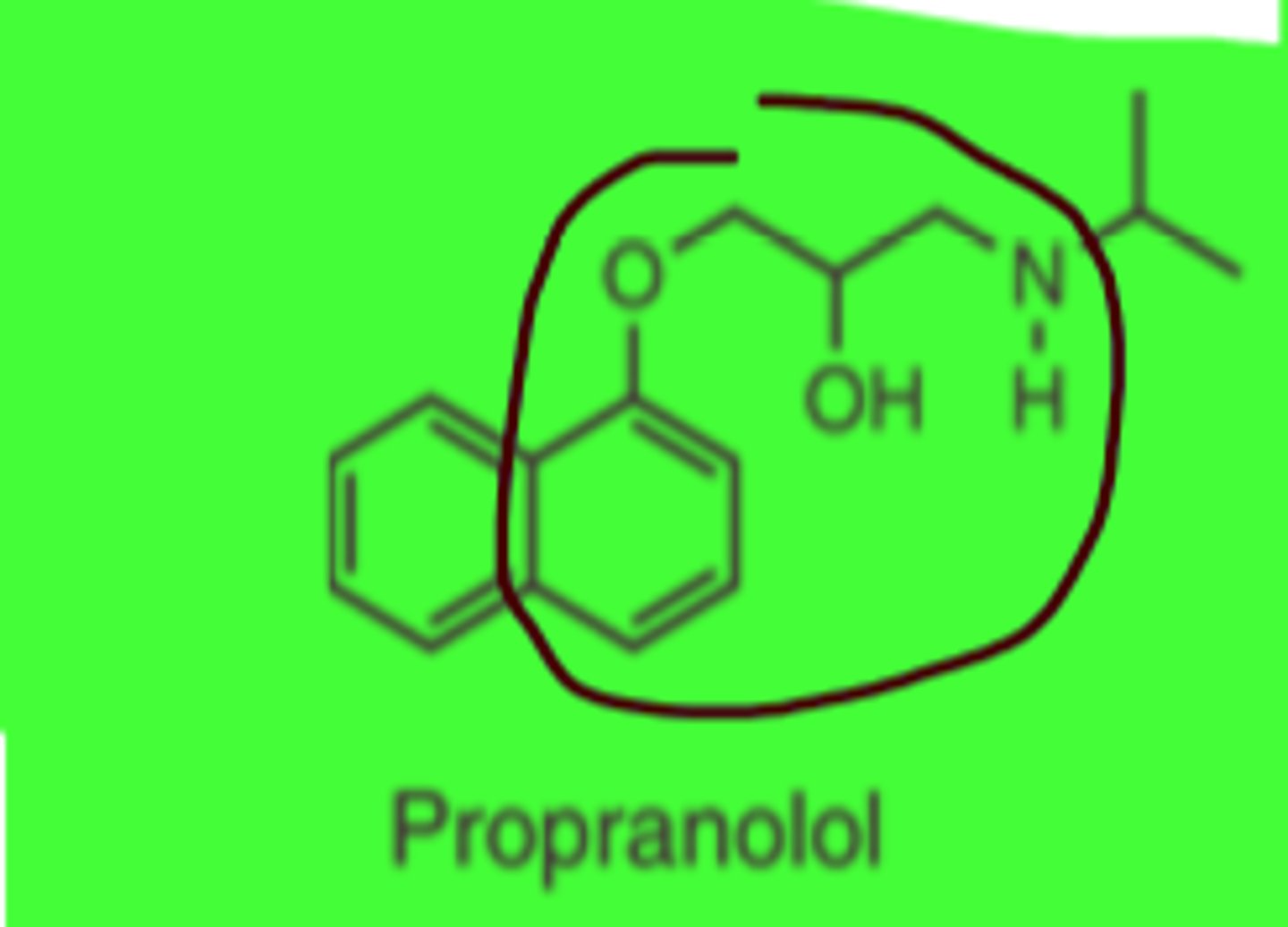
Propranolol
Phenyloxypropanolamine
(nonselective)
Sotalol
-Phenylethanolamine
-methanesulfonanilide derivative
(nonselective)
Propranolol structure
Sotalol structure
-can make a salt
-charged->can help with receptor
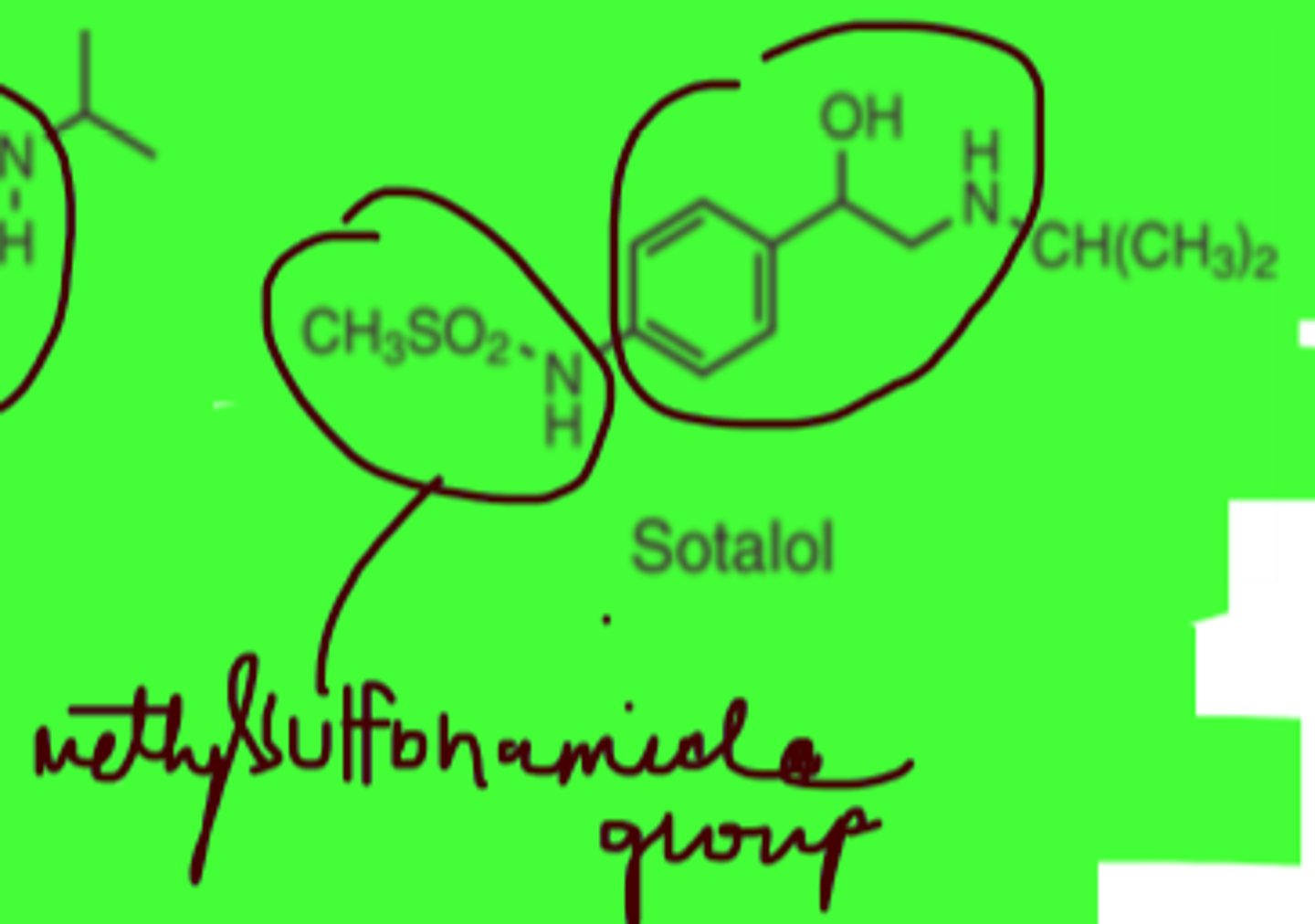
Sulfonamide is an __________ functional group
acidic
Class III Potassium Channel Blockers
Amiodarone
Dronedarone
Ibutilide
Dofetilide
Amiodarone
-also acts as class I, II, and IV anti arrhythmic
-structural analog of thyroid hormone
-aa actions & toxicity from interaction w/ nuclear thyroid hormone receptors
-highly lipophilic, eliminated slowly (several wks)
-AEs may resolve very slowly
-bioactive major metabolite: desethylamiodarone
Amiodarone structure
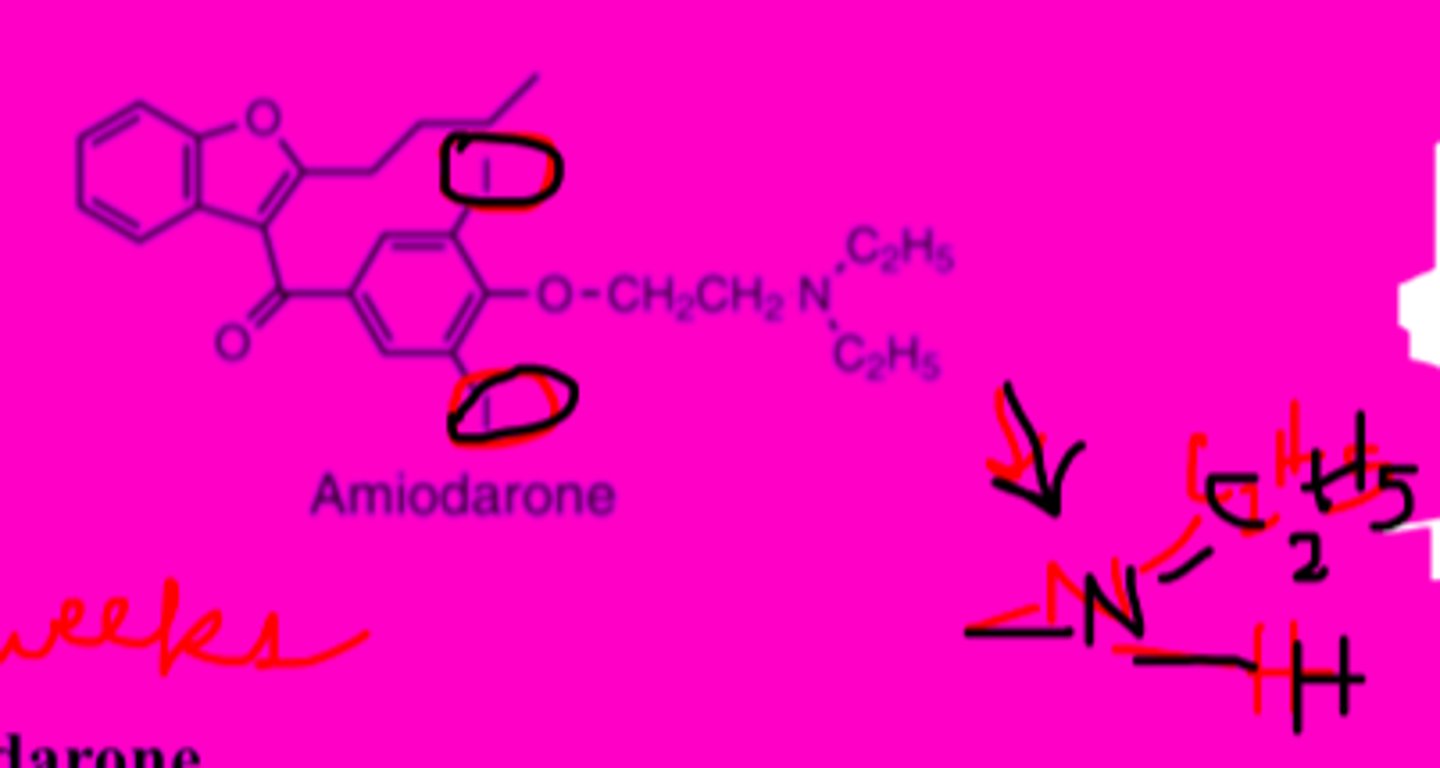
Drug interactions of amiodarone
-substrate for CYP3A4
-conc. increased by drugs that inhibit CYP3A4 (ex: histamine H2 blocker cimetidine)
-drugs that induce CYP3A4 decrease conc. (ex: rifampin)
-inhibits cytochrome P450 enzymes causing high lvls of statins, digoxin, and warfarin
Dronedarone
-benzofuran non iodinated derivative of amiodarone
-methylsufonamido group and dibutyl substitution on basic nitrogen
-less lipophilic than amiodarine
-elimination half life of 24 hrs
-oral bioavailability very poor (4%)
-high fat meal increases bioavailability (15%)
Dronedarone structure
-iodine groups of amiodarone removed to reduce toxic effects on thyroid
-methylsulfonamido group reduces lipophilicity and neurotoxic effects
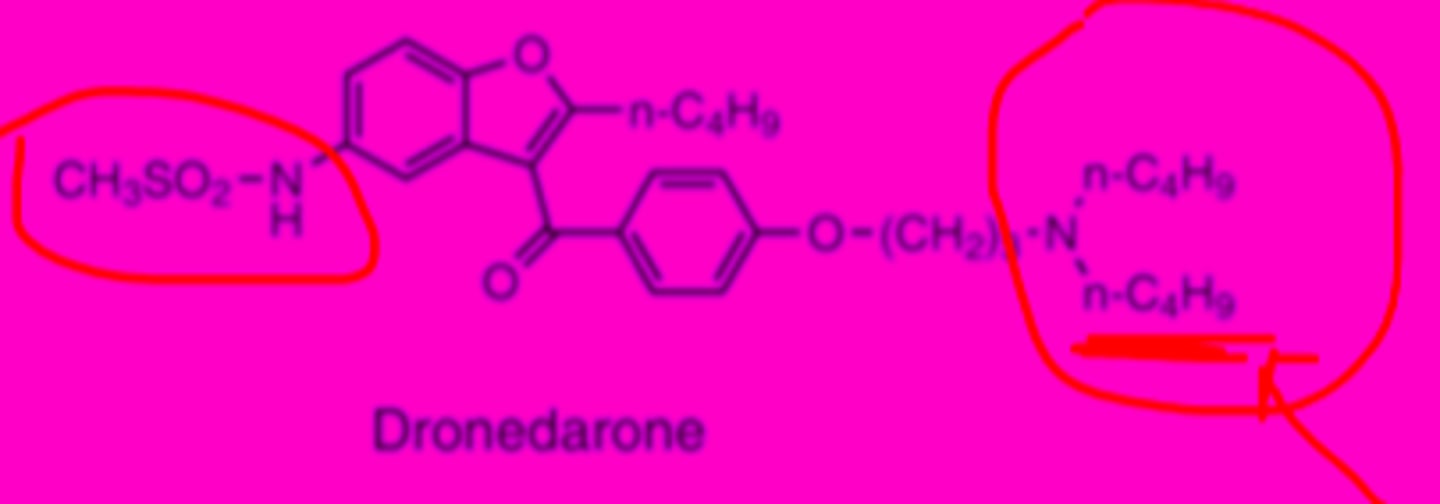
Dronedarone metabolism
after PO, metabolized by liver CYP3A4 N-debutylation into active metabolite
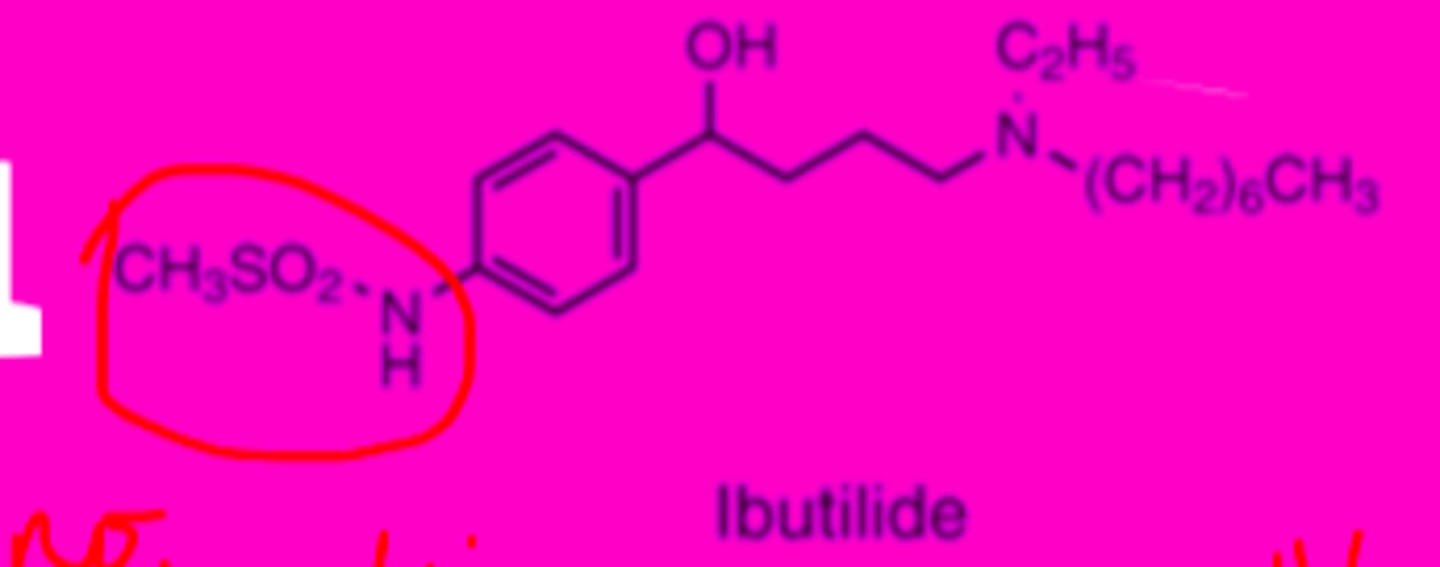
Ibutilide
-methanesulfonanilide derivative
-IV infusion only as its fumarate salt
-lacks any B-blocking activity unlike sotalol
-electrophysiologic effects characteristic of class III
Ibutilide structure
Ibutilide liver metabolism
high first pass metabolism, poor oral bioavailability
2 IV anti arrhythmic drugs
Lidocaine and Ibutilide
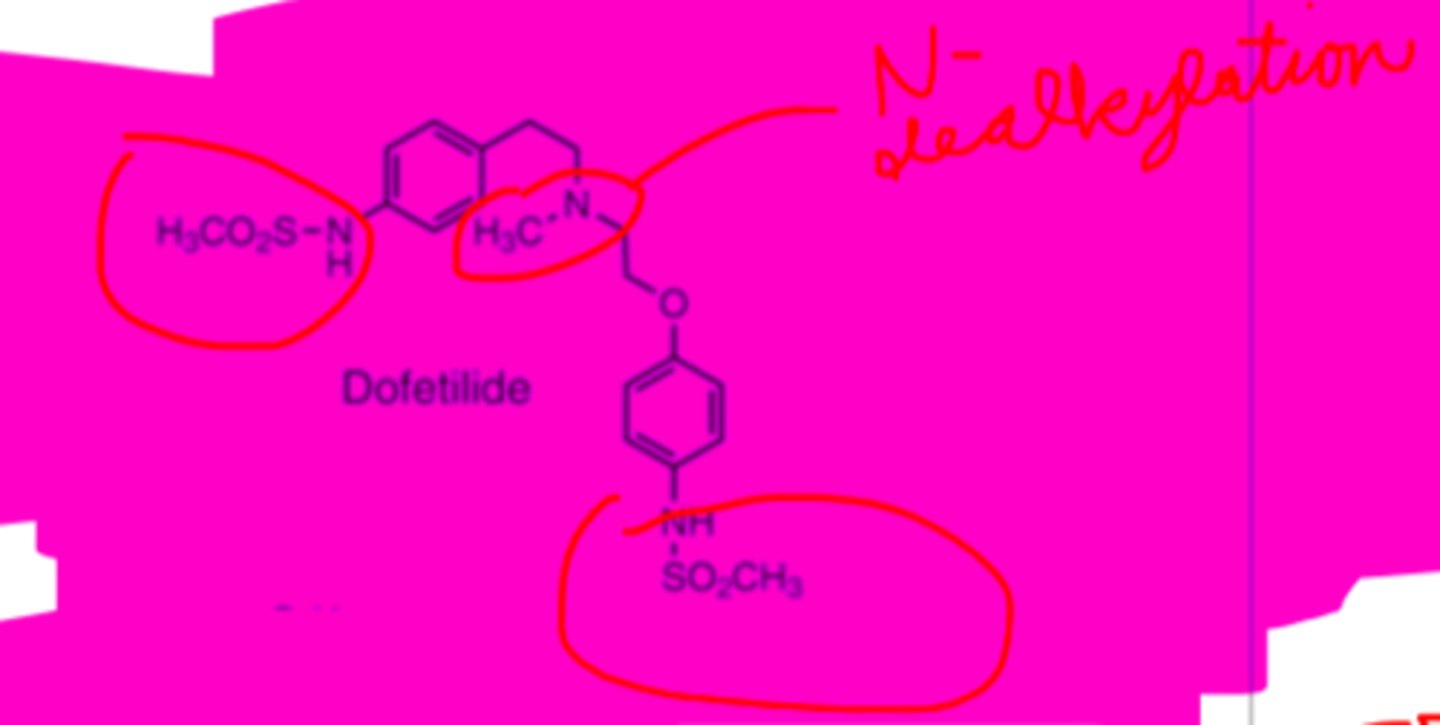
Dofetilide
-bis-methanesulfonanilide derivative
-non-B-blocking moiety of sotalol molecule
-lacks B-blocking activity
-exhibits only class III electrophysiologic effects
-more potent & selective than other class III methanesulfonanilides
Dofetilide metabolism
-hepatic CYP3A4 enzyme system via N-dealkylation & N-oxidation
-inactive or minimally active metabolites
Dofetilide structure
PO, well absorbed from GI tract (96-100% bioavailability)
Class IV Calcium Channel Blockers
Verapamil and diltiazem (non-DHPs)
dihydropyridines are less effective in cardiac tissues
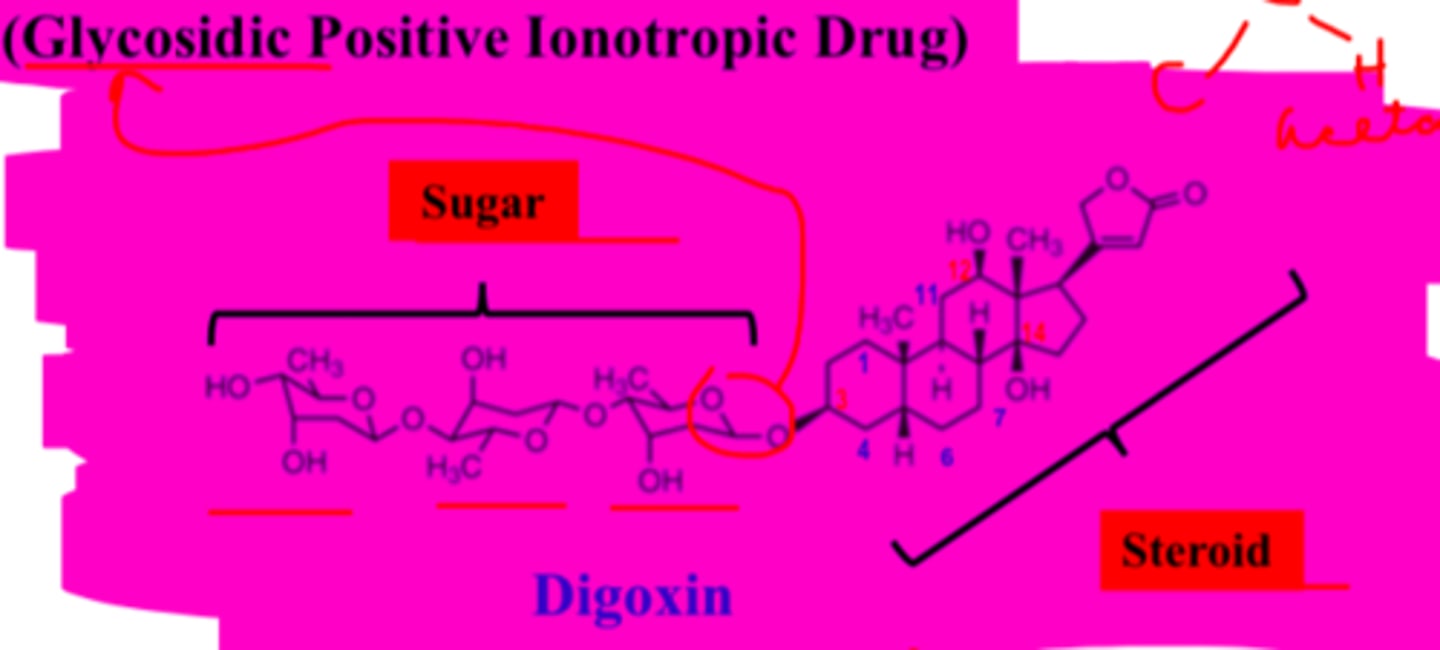
Digoxin
-cardiac glycoside or positive inotropic drug
-from digitalis purpurea or foxglove plant
-only oral positive inotropic agent
-actively secreted into urine by renal tubular cell via p-gp efflux pump
Digoxin structure
-polysaccharide (sugar) and steroid
-steroid portion has 3 hydroxyls
-C3 hydroxyl is conjugated to polysaccharide
PK and metabolism of digoxin
-oral bioavailability ranges from 70-85% of dose
-interindividual variability attributes to intestinal P-gp efflux and P-gp dependent renal elimination
-t1/2 in pts w/ normal renal function is 1.5-2 days
Basis for digoxin-drug interactions and digoxin toxicity
alterations to p-gp transport
P-gp substrates that can alter control of arrhythmias during concurrent use of cardiac glycosides (digoxin)
-antiarrhythmics
-sympathomimetics
-B-adrenergic blockers
-calcium channel blockers
Digoxin-verapamil interaction
-verapamil inhibits intestinal P-gp efflux of digoxin
-blocks intestinal secretion of digoxin into lumen
-raises digoxin blood to toxic lvls
Rifampin-digoxin interaction
-rifampin induces intestinal p-gp expression
-increases p-gp mediated secretion of digoxin
-lowers digoxin blood lvls to sub therapeutic concentrations